서 론
길랭-바레증후군(Guillain-Barré syndrome, GBS)은 감염 이후에 발생하는 급성 다발신경병(polyneuropathy)으로서 전통적으로 하지에서 상지로 진행하는 마비(ascending paralysis)를 특징으로 한다[1]. 과거에는 GBS로 진단된 환자들의 전기생리학적검사에서 탈수초신경병(demyelinating neuropathy) 소견이 흔히 관찰되었기 때문에 급성염증탈수초다발신경병(acute inflammatory demyelinating polyneuropathy, AIDP)으로 부르기도 하였으나 축삭이 주로 파괴되는 급성운동축삭신경병(acute motor axonal neuropathy, AMAN)도 GBS의 대표적인 한 형태로 알려졌다[2].
대부분의 환자에서 단상(monophasic)의 경과를 보이며 수 개월에 걸쳐 사지마비가 회복되므로 예후가 나쁘지 않은 병으로 여겨진다. 그러나 적절한 면역치료에도 불구하고 25%의 환자에서는 호흡마비와 자율신경기능장애가 동반되어 중환자실 치료가 불가피하고, 20%의 환자에서는 장기적으로 심각한 신경계 후유증이 남을 수 있다. 심지어 5% 전후의 환자에서는 사망에까지 이르기도 한다[3,4].
수 년간 이 병의 병태생리에 직접적인 역할을 하는 다양한 종류의 강글리오시드(ganglioside)와 이에 대한 자가항체의 관계가 밝혀지면서 전형적이지 않은 증상으로 나타나는 밀러피셔증후군(Miller Fisher syndrome, MFS)과 인두목위팔위약형(pharyngeal-cervical-brachial weakness type, PCB) 등을 GBS의 아형으로 분류할 수 있게 되었다[5,6]. 최근에 항-강글리오시드항체(anti-ganglioside antibody)는 진단에 도움이 되는 중요한 도구가 되었을 뿐만 아니라, 미래의 치료제 개발을 위한 핵심적인 병태생리 정보로서의 가치도 대두되고 있다[7,8].
본 론
1. 발견
1916년에 1차 세계대전에 참전하였던 프랑스의 신경과의사 Georges Charles Guillain과 Jean Alexandre Barré 그리고 전기생리의학자였던 André Strohl은 급성으로 진행하는 사지마비로 발현한 2명의 병사를 진료하였다. 당시 감염성 사지마비의 대표적인 원인으로 잘 알려진 회질척수염(poliomyelitis)과 달리 이 환자들의 뇌척수액(cerebrospinal fluid, CSF)에서는 염증세포가 없이 단백질만 증가하고(albuminocytologic dissociation, 알부민세포해리) 시간이 경과하면서 후유증 없이 회복되는 것을 확인하여 GBS 증례들을 처음으로 보고하였다[9,10].
3. 선행 감염
GBS 환자의 60-70%에서 신경계증상이 발생하기 4주 이내에(대부분의 경우 2주 이내) 상기도나 위장관 감염 등의 다양한 감염증상이 나타난다[13]. 환자-대조군 연구를 통하여 일부에서의 원인균이 밝혀졌는데 그중에서 캄필로박터제주니(Campylobacter jejuni)가 25-50%로 가장 높은 비율을 차지하였고, 거대세포바이러스(cytomegalovirus), 엡스타인-바바이러스(Epstein-Barr virus), 폐렴미코플라스마(Mycoplasma pneumoniae)가 각각 13%, 10%와 5%로 대조군에 비하여 의미 있게 높은 비율로 발견되었다[14]. 그 외에도 인플루엔자균(Haemophilus influenzae), 1형 파라인플루엔자바이러스(parainfluenza 1 virus), 인플루엔자A바이러스(influenza A virus), 인플루엔자B바이러스(influenza B virus), 아데노바이러스(adenovirus), 단순포진바이러스(herpes simplex virus), 수두대상포진바이러스(varicella-zoster virus) 등이 GBS를 일으킬 수 있는 것으로 보고되었다[14]. 최근 중남부아메리카를 포함한 25개국에서 창궐하였던 Zika바이러스가 GBS의 원인으로 보고되면서 전 세계적으로 GBS에 대한 관심이 증가하였다[15].
백신(vaccine) 접종도 GBS의 중요한 원인 중 하나로, 가장 대표적인 예는 1976년 미국에서 시행한 돼지인플루엔자(A/New Jersey/1976/H1N1)에 대한 대규모 백신 접종이다. 당시 접종을 받은 성인 100만 명 중 8.8예 미만에서 6주 이내에 GBS가 발병하였는데, 이는 같은 시기에 백신을 접종받지 않는 사람에서의 발병 빈도를 크게 상회하는 것이었다[16]. 2009년에 H1N1에 대한 대규모 백신 접종을 하면서 GBS의 발병이 증가할 것으로 우려하였으나 100만 명당 1.6예의 추가 증례가 확인되어 다른 시기에 인플루엔자 접종을 받은 경우와 비슷한 양상으로 나타났다[17]. 과거 GBS를 앓았던 환자에서 백신 접종을 하는 것이 안전한지에 대한 논란은 지속되고 있다. 이에 대한 해답을 제시하기는 어렵지만 인플루엔자의 감염 또한 GBS의 원인 중 하나이며, 인플루엔자에 감염되어 GBS가 발병할 확률은 백신 접종에 의하여 발병할 확률보다 4-7배 정도 높을 것으로 추정되고 있다. 따라서 일부 가이드라인에서는 최근 3개월 이내 GBS를 앓거나 백신에 의한 GBS를 앓은 병력이 있는 환자를 제외하고 백신 접종은 상대적으로 안전하다고 제안하고 있으며, 현재까지 GBS에서 호전된 환자에서 백신 접종 이후 GBS가 재발한 사례는 보고되지 않았다[18].
4. 병리기전
AIDP의 병리기전에 대한 연구는 주로 사후 부검과 동물모델을 통하여 이루어졌다. 사망한 환자의 척수신경뿌리(spinal root)와 운동 및 감각신경에서 T세포와 대식세포(macrophage)가 광범위하게 침윤되었고 분절탈수초현상(segmental demyelination) 및 이차적인 축삭변성(axonal degeneration)이 관찰되었다[19]. 이후 현재까지 병을 일으키는 원인 항체는 알려지지 않았으나 활성화된 보체(complement)가 작용하는 것을 발견하여 슈반세포(Schwann cell)표면에 병을 일으키는 미상의 항체가 결합하고 여기에 보체가 결합하여 수초를 파괴하는 것으로 추측하고 있다[20]. 수초가 손상되면 1주일 이내에 대식세포가 나타나 이를 제거한다. T세포의 역할은 아직 잘 모르지만 병을 유도하는 초기 단계에 작용할 것으로 추측하고 있다[21].
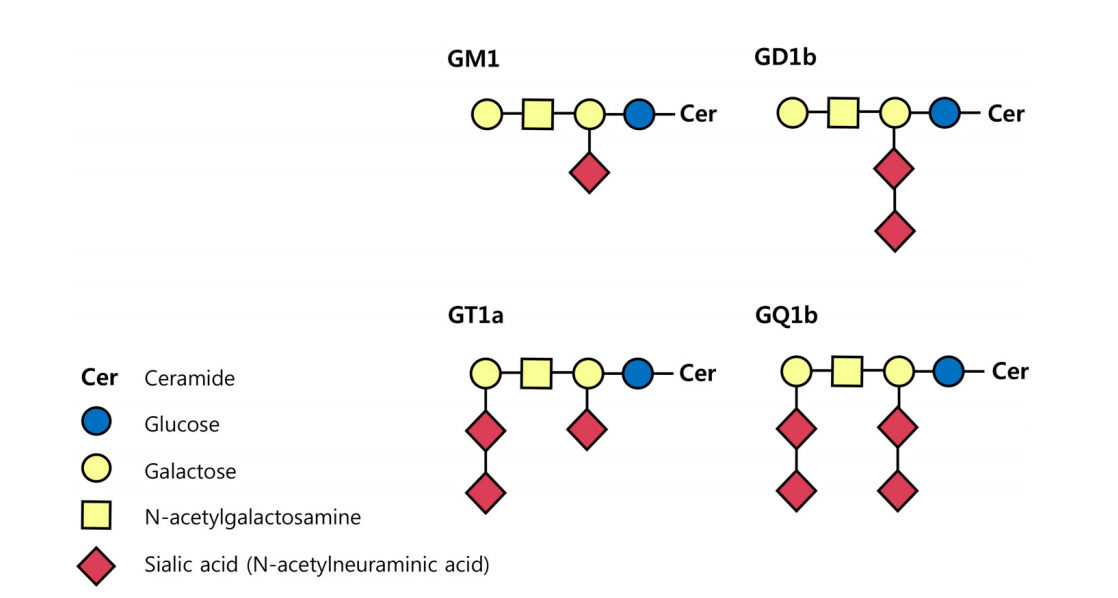
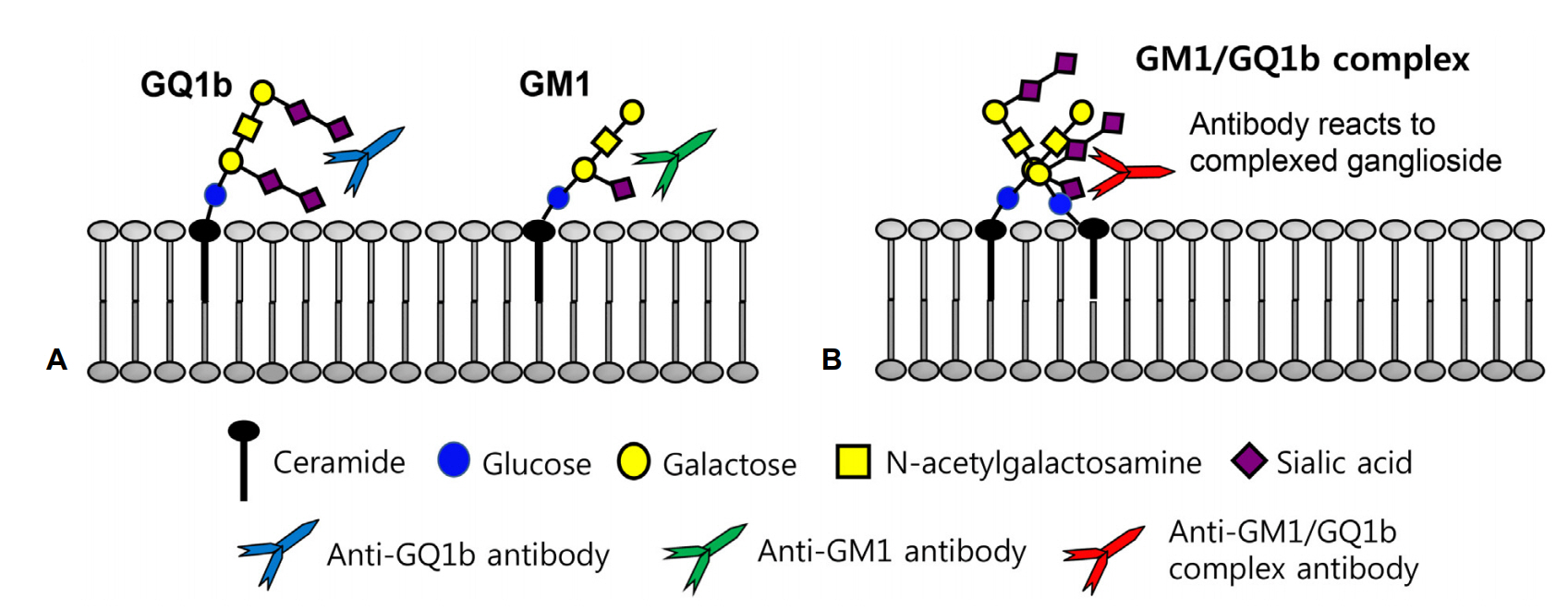
AMAN과 MFS는 항-강글리오시드항체(anti-ganglioside antibody)와 분자모방(molecular mimicry) 이론을 통하여 상대적으로 많은 병리기전이 밝혀졌다. 강글리오시드는 세라미드(ceramide)를 기반으로 올리고당(oligosaccharide)에 하나 또는 그 이상의 육탄당(hexose)과 시알산(sialic acid)이 결합된 형태로 신경막을 구성하는 중요한 요소 중 하나이다. 시알산의 위치와 수에 따라 이름이 결정되는데, 대표적인 4종의 강글리오시드인 GM1, GD1b, GT1a, GQ1b의 M, D, T, Q는 각각 mono-, di-, tri- 그리고 quadri-sialosyl 그룹을 의미한다(Fig. 1) [4]. 분자모방이론은 감염에 의하여 유발된 강글리오시드에 대한 자가항체가 어떻게 GBS의 중요한 병인으로 작용하는지 설명해 준다. 지질소당류(lipo-oligosaccharide, LOS)는 GBS의 대표적 원인균인 C. jejuni의 바깥막(outer membrane)을 구성하는 구성 성분이다. Penner O:19 혈청형(serotype)을 가지는 C. jejuni는 강글리오시드 GM1 또는 GD1a와 유사한 형태의 LOS가 있어 이 혈청형의 C. jejuni에 대한 자가항체는 AMAN을 유발하고, Penner O:10 혈청형을 가지는 C. jejuni는 강글리오시드 GT1a 또는 GQ1b와 유사한 형태의 LOS가 세포 표면에 있어 MFS나 MFS의 유사 아형을 일으킨다[22,23]. 그 외에 MFS 환자에서 동정한 인플루엔자균에서도 GQ1b 강글리오시드와 유사한 LOS가 확인되었다[24].
강글리오시드 GM1과 GD1a에 대한 immunoglobulin (Ig) G 자가항체는 AMAN뿐만 아니라 급성운동전도차단신경병(acute motor conduction block neuropathy, AMCBN)과 일부의 급성운동감각축삭신경병(acute motor and sensory axonal neuropathy, AMSAN) 환자에서도 관찰된다(Table 1). GQ1b에 대한 IgG 자가항체는 Bickerstaff뇌간뇌염(Bickerstaff’s brainstem encephalitis, BBE)에서도 관찰된다[25]. GT1a에 대한 IgG 자가항체는 PCB의 발생과 밀접한 관계가 있으며 GQ1b 강글리오시드와 교차반응(cross reaction)이 일어날 수 있다[26]. 강글리오시드 GQ1b는 눈근육운동신경이나 근육방추(muscle spindle)와 관계가 있고 GT1a는 혀인두신경(glossopharyngeal nerve)이나 미주신경(vagus nerve)과 더 관계가 있는 것으로 보인다. 강글리오시드 GM2에 대한 IgM 및 IgG 자가항체는 거대세포바이러스에 의한 GBS와 관계가 있고 주로 AIDP 형태로 발생하지만 AMSAN이나 양측성안면신경마비(facial diplegia)에서도 발견되는 등 다양한 임상양상을 보였다[27].
최근 Kaida 등[28]은 단일항체뿐만 아니라 두 개의 강글리오시드가 결합한 형태의 강글리오시드복합체(ganglioside complexes)에 대한 자가항체가 환자의 혈청에 존재하고 소수에서는 단일항원에는 반응이 없으나 강글리오시드복합체에만 반응하는 GBS 환자가 있음을 알아냈고, 그 이후로 다양한 종류의 항-강글리오시드복합항체와 임상적 인과관계가 보고되고 있다(Table 1). 일부 항-강글리오시드 복합항체는 항체특이적인 임상증상을 보이는데 그 예로 강글리오시드GD1a/GD1b복합항체를 가진 환자는 75%에서 삼킴장애를 보였고 50%에서 호흡마비로 인한 기계호흡이 필요하였다. 국내에서도 강글리오시드복합체 GM1/GT1a와 GM1/GQ1b에 대한 IgG 자가항체가 강양성을 보인 환자에서 호흡마비를 동반한 MFS-급성숨뇌마비(acute bulbar palsy)중복증후군이 보고된 바 있다(Fig. 2) [29].
5. 임상양상
전형적인 증상은 빠르게 진행하는 사지근력저하로 감각이상과 뇌신경기능장애가 흔하게 동반된다. 하지에서 시작하여 상지로 진행하는 근력저하를 보이지만 더 근위부에서 증상이 시작되거나 상지마비 없이 하지마비로만 증상이 발생하여 초기에 척수병증으로 오인되기도 한다[30]. 감각증상은 주로 무감각(numbness)이나 감각이상(paresthesia)으로 나타나며 그 빈도나 정도는 GBS의 아형에 따라 다양하다[1]. 많은 환자(54-89%)에서 통증을 동반한 이상감각, 허리 통증, 근육통 그리고 수막자극증(meningism) 양상의 통증을 호소하는데, 그중 30-40%는 사지마비가 진행하기 이전에 통증이 선행하기도 한다[31]. 약 50%의 환자에서 뇌신경장애를 동반하며 얼굴신경마비, 눈근육마비 그리고 숨뇌마비(bulbar palsy) 순서로 흔하다[1].
처음 증상이 시작하여 가장 심하게 되는 시점(nadir)에 이르기까지 짧게는 12시간에서 최장 4주가 소요되며 80%의 환자에서 2주 이내에 도달한다[32]. 이후 정점지속(plateau) 상태로 머물다가 호전되기 시작하는데 정점지속의 기간은 2일에서 6개월까지 매우 다양하게 보고되고 있다[33].
1) 급성염증탈수초다발신경병(AIDP), 급성운동축삭신경병(AMAN), 급성운동감각축삭신경병(AMSAN) 및 급성운동차단신경병(AMCBN)
가장 전형적인 GBS의 발병 양상은 진행하는 사지마비의 형태이며 이에 해당하는 것이 AIDP, AMAN 및 AMSAN이다. 1990년대 초까지 AIDP는 GBS와 구분 없이 동일한 용어로 혼용해서 사용해 왔다. 이는 GBS를 전기생리학적으로 연구하였던 서양에서 대부분의 GBS가 탈수초변화(demyelinating change)를 보였던 것에 기반한다. GBS 환자의 신경전도검사에서 관찰되는 축삭손상은 신경을 감싸는 슈반세포와 수초의 심한 탈락에 의한 이차적인 변화로 여겼다[34]. 그러나 AMAN과 연관한 병리기전이 밝혀지고, 특히 C. jejuni 감염과 연관한 항-강글리오시드항체가 발견되면서 AMAN은 축삭집(axolemma)이 주된 자가면역의 공격 대상이 되어 축삭손상이 야기되는 독특한 GBS의 한 아형으로 받아들여졌다[34].
AIDP는 감각과 운동신경을 동시에 침범하며 뇌신경장애, 자율신경이상 및 통증이 비교적 흔하게 동반되고 신경전도검사에서 탈수초이상이 보인다. AMAN은 순수한 운동신경병으로 임상양상과 신경전도검사에서 감각신경기능은 정상 소견을 보이나 10% 미만에서 감각증상을 호소하기도 한다[2,35]. AMAN에서도 통증은 흔하게 동반되나 뇌신경장애와 자율신경기능장애의 빈도는 AIDP와 비교하여 높지 않다[2,4,13,36]. AMAN은 신경축삭이 주된 면역 공격의 대상이 되기 때문에 조기에 Waller변성(Wallerian degeneration)이 동반되면 병의 진행이 빠르고 회복이 더딘 경우가 많다. 감각신경이상을 동반한 축삭신경병으로서 드물게 관찰되는 아형인 AMSAN이 있는데, GBS의 또 다른 아형으로 분류하기도 하지만 AMAN의 특정 단계에서 경미한 감각신경이상이 동반된 상태로 여겨지기도 한다[37]. 북미와 유럽을 포함한 서양에서의 GBS는 대부분 AIDP의 형태로 발병하고 AMAN은 매우 드물어서 5% 전후로 추정되지만 아시아와 중남미에서는 상대적으로 AMAN 환자가 30-56%의 높은 비율로 보고되어 지역에 따른 분명한 차이가 알려져 있다[2,12,13,38].
전기생리학적 연구를 바탕으로 한 국내 연구에서 신경전도검사의 기준에 따른 AIDP와 AMAN의 비율은 일관되지 않은 결과를 보이는데, 이는 탈수초를 판단하는 기준의 차이에서도 기인하지만 일부의 AMAN 환자들은 질병의 초기에 두드러진 운동전도차단(motor conduction block)을 보일 수 있기 때문에 기존의 전기생리학적 기준으로는 AIDP로 분류되었을 가능성이 있다[13,39,40]. 2003년 Capasso 등[41]은 항-강글리오시드항체가 양성이면서 감각이상은 동반하지 않은 AMAN을 시사하는 환자에서 신경전도검사에서는 두드러진 가역적인 운동전도차단을 보일 수 있음을 관찰하고, AMAN의 병태생리를 가지는 경우에도 전기생리검사에서는 탈수초신경병으로 분류될 수 있음을 제시하며 AMCBN으로 명명하였다. 이후 이 현상은 동물모델을 이용한 연구들과 환자의 항체결과를 포함한 전기생리학 연구를 통하여 원인이 규명되었다. 즉, 항-강글리오시드항체가 결절주변축삭막(paranodal axolemma)에 존재하는 강글리오시드를 공격할 때 심한 축삭퇴행(axonal degeneration)과 Waller변성으로 진행하는 경우 전형적인 AMAN의 양상으로 발전하는 반면, 항-강글리오시드항체가 결절주변수초고리(paranodal myelin loop)의 분리를 야기하였다가 더 이상 진행하지 않고 빠른 시간 내에 회복하면 AMCBN으로 될 수 있다는 것이 밝혀졌다[42-44].
2) 밀러피셔증후군(MFS)
1956년에 Charles Miller Fisher는 전체외안근마비(total external ophthalmoplegia), 심한 실조(ataxia) 및 무반사(areflexia) 증상이 급성으로 나타난 세 명의 환자를 경험하였고 그중 한 환자의 CSF에서 알부민세포해리를 확인하였다. 무반사와 알부민세포해리는 GBS의 특징적인 소견으로 위 환자들을 GBS의 아형 중 하나로 제안하였다[45]. 이후 유사한 많은 환자들이 보고되었고 위의 세 징후를 가진 급성 신경염을 피셔증후군(Fisher syndrome) 또는 MFS이라고 불렀다. 이와 더불어 의식저하와 척수로(spinal tract) 증상을 가지는 BBE, 사지마비가 진행하는 MFS-GBS중복증후군(overlapping syndrome), 다양한 다른 뇌신경증상 또는 감각이상증상을 동반하거나 항-GQ1b 강글리오시드항체가 양성이면서 세 징후 중 일부가 양성인 다양한 변이형이 존재한다[46,47].
MFS을 이해하는 데는 1992년 발견된 항-GQ1b 강글리오시드항체가 중요한 역할을 하였다[5]. 항-GQ1b 강글리오시드항체는 MFS 환자 급성기 혈청의 약 70-90%에서 발견된다[48]. 세 징후 중에서 외안근마비와 다른 두 징후 중 하나만 동반하는 경우 기존의 MFS의 진단기준에는 맞지 않으나 항-GQ1b 강글리오시드항체가 양성이면 항-GQ1b항체증후군(anti-GQ1b antibody syndrome)으로 부르기도 하여 항체를 통한 판단이 질병의 병태생리를 이해하고 분류하는데 도움을 주었다[49,50].
MFS의 세 징후 중 가장 중요하고 일관성 있는 증상인 외안근마비는 다양한 양상으로 나타날 수 있다. 증상이 최고조에 이르렀을 때는 양측성 완전마비가 될 수 있으나 불완전마비가 되거나 비대칭적 외안근마비로 나타나기도 하며 외안근 중에서 가장 두드러진 마비를 동반하는 근육은 외직근(lateral rectus muscle)이다[51]. 상대적으로 드물지만 양측성 외전신경마비(abducens palsy), 순수 내안근마비(internal ophthalmoplegia) 등의 비특이적 국소 증상으로 나타나기도 한다[52,53]. 다양한 종류의 감염 이후에 발병하지만 실제 원인균이 밝혀지는 경우는 드물고 약 20%의 환자에서 C. Jejuni와 8%의 환자에서는 H. Influenzae와 인과관계가 있음이 알려졌다[4].
3) 그 외 아형들
GBS의 진단기준을 만족하지는 못하지만 순수한 감각신경증상으로만 나타나는 급성감각실조신경병(acute sensory ataxic neuropathy, ASAN) 또한 GBS의 아형 중 하나로 분류되고 있다[54]. 이 환자들 중 일부에서 항-GD1b 강글리오시드항체가 발견되어 GBS와 유사한 병태생리를 공유한다는 사실을 알게 되었고 GD1b 강글리오시드를 면역유발제로 사용하여 만든 ASAN 동물모델을 통하여 증명되었다[54,55].
그 외에도 각종 항-강글리오시드항체나 CSF검사에서 보이는 알부민세포해리 소견을 통하여 GBS의 국소적인 변이형(regional variant)으로 분류되는 다양한 아형이 존재한다. 그 대표적인 예로 항-GT1a 강글리오시드항체가 양성이면서 삼킴장애, 목근육 위약 및 상지 근위부 근력저하를 특징으로 하는 PCB가 있다[6]. 최근 MFS과 PCB의 중간 단계에 해당하는 급성숨뇌마비변이형(acute bulbar palsy variant)이 보고되었는데, 이 증례들에서도 항-GT1a 강글리오시드항체가 일관성 있게 발견되었다[56]. 항-GD1a 강글리오시드항체는 항-GM1 강글리오시드항체와 더불어 AMAN을 유발하는 대표적인 인자로 알려져 있지만, 양측안면신경마비와 연관성이 높을 것으로 추정한다[13,57]. 이 외에도 사지의 감각이상을 동반한 양측안면신경마비(facial diplegia with limb paresthesia)도 드문 GBS의 아형으로 알려졌다[58,59].
4) 자율신경기능이상
자율신경의 이상은 GBS환자의 60-70%에서 동반되는 흔한 신경계 합병증이며 혈압의 심한 변동, 부정맥, 혈관운동(vasomotor) 신경 이상 그리고 위장운동장애가 대표적인 예이다. 이는 GBS의 연속선상에서 자율신경의 탈수초현상에 의하여 발생하는 것으로 생각된다[60]. 치료가 필요하지 않은 경우가 대부분이지만, 고혈압이 발생한 환자의 약 11%에서는 만성적인 고혈압으로 이어지며 일부의 환자에서는 심각한 고혈압으로 인하여 사망하기도 한다[60]. 일부 환자에서는 가역적후뇌병(posterior reversible encephalopathy syndrome)이 보고되었는데 상당수가 면역글로불린의 사용과 관련하여 치료 중 발생하였다[61]. 가장 심각한 합병증은 동정지(sinus arrest)로 항GQ1b항체와 관련하여 발생할 수 있음이 보고되었으나 일반적으로 심한 자율신경이상은 AIDP에서 더 흔하다[62,63]. 자율신경이상이 있는 환자의 약 7%에서 사망할 수 있고 초기에는 증상이 없는 경우가 많으므로 이를 예방하기 위해서는 심혈관계와 호흡기계를 꾸준히 감시하는 것이 중요하다[60]. 일반적으로 급성기가 지나면서 호전되어 사지마비 증상보다 빨리 회복되지만 기립저혈압 증상은 회복기에도 지속될 수 있다[10].
6. 진단
GBS의 진단기준은 1978년에 미국 National Institute of Neurological Disorders and Stroke (NINDS)에서 1976년과 1977년에 걸쳐 발생한 GBS에 대하여 돼지인플루엔자백신(swine influenza vaccine)과의 관계를 조사하면서 체계화되기 시작하였고 1981년에 처음으로 만들어졌다. 현재 가장 많이 사용되는 Asbury와 Cornblath가 발표한 진단기준은 1990년에 이를 보완한 것으로 진행하는 사지마비증상과 심건반사의 소실(또는 저하)을 만족하면서 다른 신경계증상 또는 징후와 검사실 소견을 통하여 판단하도록 하였다[1]. 2011년에 발표된 Brighton기준 역시 2009-2010년 사이에 접종된 H1N1 돼지 인플루엔자와 GBS 발생 간의 관계를 조사하기 위하여 만들어졌다[64]. 이 진단기준들은 예방접종의 안전성을 밝히기 위하여 만들어진 것들로 전형적인 형태의 GBS를 진단할 때만 적용이 가능하여 민감도가 낮다는 단점이 있다. Brighton기준은 GBS의 가능성이 높은 수준에 따라 4단계로 분류하며, 대칭적인 사지마비와 심건반사의 감소(또는 소실) 및 일회성의 진행양상 등 전형적인 임상양상을 보이고 뇌척수액의 알부민세포해리와 신경전도검사의 이상을 보이는 1수준(level 1), 전형적인 임상증상을 보이지만 뇌척수액의 알부민세포해리와 이상 신경전도검사 소견 중 하나만 만족하는 2수준(level 2), 전형적인 임상증상만 보이고 뇌척수액검사와 신경전도검사에서는 뒷받침되지 않는 3수준(level 3), 임상증상도 전형적이지 않지만 GBS가 아닌 다른 원인으로 증상을 설명할 수 없는 경우를 4수준(level 4)으로 정의하였다. 그 결과, AIDP가 많은 네덜란드 그룹에서 이를 적용하면 전체 GBS 환자 중 61%가 1수준으로 진단되었고 4수준은 6%에 불과하였으나 한국 환자들에게 적용하였을 때는 겨우 9% 환자에서 1수준으로 진단되었고 24%의 환자가 4수준으로 진단되었다[33,65]. 이러한 단점을 보완하기 위하여 2014년에 GBS Classification Group은 임상증상을 기준으로 한 새로운 분류 기준을 제시하였고(Table 2), 이 기준은 다양한 국소변이형으로 나타날 수 있는 아시아 지역 환자들을 분류하는데 도움이 되었다[3,66].
1) 뇌척수액검사
뇌척수액검사에서 보이는 알부민세포해리는 GBS를 진단하는데 중요한 검사 소견으로 알려져 있다. 그러나 대부분의 환자에서 처음에는 정상 소견을 보이다가 사지마비가 발생한지 3일 이상 경과한 뒤에야 50%의 환자에서 알부민세포해리가 관찰되므로 초기의 감별진단에는 도움이 되지 않을 수 있다[33]. AIDP에서는 뇌척수액의 단백상승 소견이 비교적 흔하게 나타나지만 AMAN이나 일부 국소변이형 GBS에서는 두드러지지 않는 경향을 보이므로 결과를 해석할 때 이를 고려해야 하며 진단을 위한 반복적인 뇌척수액검사는 권장하지 않는다[13,18].
2) 신경전도검사
전통적으로 신경전도검사(nerve conduction study)는 GBS를 진단하는데 매우 중요한 도구로 사용되어 왔다[9]. 이를 기반으로 탈수초신경병의 판단 기준이 발달하였고, GBS의 아형을 구별하고 장기적인 예후를 예측하는데 도움을 주었다[67]. 진단율을 높이기 위해서는 최소 4군데 이상의 운동신경과 3군데 이상의 감각신경, F파 그리고 H반사를 확인해야 한다.
AIDP에서 보이는 신경전도검사의 특징적인 소견은 운동신경의 종말잠복기(terminal latency)의 연장, 신경전도속도의 감소, F파의 지연, 비정상적으로 연장된 시간분산(temporal dispersion) 및 전도차단(conduction block) 등이 있다. 상대적으로 비복신경(sural nerve)이 보존되는 현상이 종종 관찰되어 다른 염증성 또는 탈수초신경병과 감별되는 특징으로 여기기도 한다[68,69]. AMAN에서 보이는 신경전도검사의 특징은 운동신경의 신경전도속도는 보존되면서 복합근육활동전위(compound muscle action potential)의 진폭이 저하되는 것이다.
GBS 환자에서 신경전도검사를 시행할 때 몇 가지 고려해야 하는 중요한 점이 있다. 우선, 질병의 초기에 시행한 신경전도검사에서는 정상 소견을 보이거나 경미한 이상만 나타날 수 있으며, 일반적으로 사지마비가 생기고 2주 이상 경과한 뒤에야 전형적인 이상을 관찰할 수 있다[38]. 따라서 시간 경과에 따라 반복적으로 검사를 시행하여 변화의 추이를 판단하는 것이 중요하다. 둘째, 신경전도검사를 통한 AIDP와 AMAN의 감별이다. 기존의 전기생리학적 분류기준에서 GBS와 AIDP는 같은 질환으로 여겨졌기 때문에 AMAN으로 분류하는 것에 더 인색한 경향이 있다[70]. 특히 가장 널리 쓰이는 Hadden의 기준은 전도차단을 탈수초의 중요인자로 포함시켜서 이 기준을 적용하였을 때 대부분의 GBS가 AIDP 아형인 서양에서는 큰 무리가 없지만, AMAN이 상당수를 차지하는 아시아 지역에서는 AMAN의 발생률이 저평가되는 문제가 발생한다[13,38]. 특히, 경미한 증상으로 나타나거나 초기에 검사를 시행한 AMAN 환자들 중 일부는 가역적 전도차단과 종말잠복기의 연장만 보이다가 빠르게 회복되는 경우가 있어 전기생리학적으로는 AMCBN으로 별도로 분류할 필요가 있으며, 이들은 병태생리적으로는 AMAN과 같거나 유사한 기전에 의하여 발생하지만 상대적으로 좋은 예후를 보인다[12]. 위와 같은 연유로 좀 더 이른 시기에 효율적으로 AMAN과 AIDP를 구분하기 위하여 다양한 노력들이 시도되었는데, 신경전도검사에서 특정 부위 및 종류에서의 특징을 찾아내거나, 신경흥분성검사(nerve excitability test)와 같은 새로운 기술을 이용하는 방법들이 그 예이다[68,71,72].
7. 치료
GBS가 진단되면 최대한 빠른 시기에 치료를 시작해야 추가적인 신경손상을 예방하고 장기적인 예후에 도움이 될 수 있으며 이는 뇌혈관질환에서 말하는 ‘Time is brain’과 유사한 개념으로 생각할 수 있다[73]. 현재까지 무작위대조시험(randomized controlled trial)을 통하여 효과가 검증된 치료제는 면역글로불린정맥주사(intravenous immunoglobulin)와 혈장분리교환술(plasmapheresis)이 있다[74]. 면역글로불린은 총 2 g/kg를 투약하는데 2일(1 g/kg/day)에 나누어 주거나 또는 5일(0.4 g/kg/day)에 나누어 줄 수 있다. 두 가지 방법의 효과에는 차이가 없고 빠른 속도로 면역글로불린을 투여한 환자에서 치료와 연관된 변동(treatment related fluctuation)이 더 자주 발생한다는 보고가 있어 5일에 걸쳐 주사하는 것이 일반적이다[75]. 혈장분리교환술은 2주 동안 총 5회에 걸쳐 시행하고 1회에 2-3 L의 혈장을 분리교환한다. 두 가지 치료 모두 사지마비가 발생하고 2주 이내에 치료를 시작하는 것이 더 효과적이다[18,73]. 면역글로불린정맥치료와 혈장분리교환술을 병행하는 것은 두 치료를 단독으로 시행하는 것과 비교하여 효과에서 유의한 차이를 보이지 않았다[76]. 경구 또는 정맥주사를 통한 스테로이드는 대증치료와 효과의 차이가 없었고 일부 연구에서는 오히려 증상이 악화되었다[77]. 그 이유는 스테로이드가 대식세포의 제거자(scavenger)로서의 역할을 억제시켜 신경재생이 지연되기 때문으로 생각된다[78].
많은 환자들이 면역글로불린과 혈장분리교환술로 치료를 받은 이후에도 오랜 기간 동안 사지마비가 지속되거나 심한 신경후유증이 남게 되므로 더 나은 치료를 위한 연구가 진행되고 있다. 현재 네덜란드 그룹에서 modified Erasmus GBS outcome scale (mEGOS)을 통하여 예후가 나쁠 것으로 예상되는 환자들을 대상으로 2차 면역글로불린을 투약하는 무작위대조시험을 진행 중이며 그 결과는 2019년에 발표될 예정이다(SID-GBS RCT trial) [79].
1) 일반적인 치료
GBS는 급성기에 적절한 치료를 하면 상당수의 환자에서 회복을 보이므로 대증치료를 통한 합병증의 예방 또한 매우 중요하다. 특히 호흡저하로 인한 기계환기가 필요할 수 있으므로 증상이 악화되는 시기에는 집중치료실에서 치료하는 것이 안전하다. 호흡기능을 판단하는데 폐활량(vital capacity)이 20 mL/kg 미만으로 감소, 최대흡기압(maximum inspiratory pressure)과 최대호기압(maximum expiratory pressure)이 각각 30 cmH2O, 40 cmH2O 미만으로 감소하면 기관내삽관(intubation)을 고려해야 한다[10]. 그 외에도 심박동이 분당 100회 이상의 빈맥을 보이거나 목빗근(sternocleidomastoid muscle)이나 사각근(scalene muscle)과 같은 호흡보조근을 사용하여 호흡하는 경우에도 주의해야 한다. 반대로 기계환기 이후 호흡기계를 제거하는 지표(weaning)로는 폐활량이 15 mL/kg 이상으로 회복되는 경과이거나 최대흡기압이 30 cmH2O 이상으로 회복하면 고려할 수 있으며, 장기적인 기관내삽관으로 인한 합병증을 예방하기 위하여 기관내삽관의 기간은 최소화해야 한다[10]. 그 외에도 심혈관계의 이상으로 인한 동정지가 발생할 수 있으므로 초기부터 심혈관계 감시를 시작하여 필요한 경우 임시박동조율기(temporary pacemaker)를 삽입해야 하고, 비뇨기계 및 소화기계 이상을 포함한 다양한 자율신경기능장애 유무를 확인해야 한다.
폐렴이나 요로감염 등의 새로운 감염이 발생하지 않도록 주의가 필요하며 경련통(cramping pain)을 포함한 다양한 통증에는 비스테로이드소염제, 가바펜틴(gabapentin), 프레가발린(pregabalin) 및 카르바마제핀(carbamazepine) 등을 사용하여 증상을 경감시킬 수 있다. 재활치료와 정신적 지지 또한 중요하다[10].
2) 치료와 연관된 변동
면역글로불린정맥치료나 혈장분리교환술을 받은 10%의 환자에서 처음에는 증상의 호전을 보이다가 다시 증상이 악화되는 경과를 보일 수 있는데, 이를 치료와 연관된 변동(treatment related fluctuation, TRF)이라고 한다[74]. 주로 치료를 시작하고 8주 이내에 나타나며 투여하였던 면역글로불린의 혈중농도가 저하되거나 혈장분리교환술의 효과가 없어질 시기가 되어서도 지속되는 자가면역작용이 신경을 공격하여 발생하는 것으로 추정한다. 반복적인 면역치료가 도움이 되지만 이에 대한 무작위대조연구는 없다[18].
일부 GBS로 진단된 환자는 반복적인 증상의 악화를 경험하는데 증상의 진행이 4주 이상 계속되는 경우는 반드시 만성염증탈수초다발신경병(chronic inflammatory demyelinating polyneuropathy, CIDP) 가능성을 고려해야 하며 한 전향연구(prospective study)에 의하면 GBS로 처음 진단된 환자의 5%가 추후에 급성으로 발생한 CIDP (acute onset CIDP, A-CIDP)로 다시 진단되었다[36]. 몇 가지 특징이 GBS-TRF와 A-CIDP를 감별할 수 있도록 해 주는데, 선행 감염이 뚜렷한 경우 안면근육의 위약, 자율신경이상 또는 호흡근의 위약을 동반한 경우에는 A-CIDP보다는 GBS를 더 시사한다고 할 수 있다[80]. 이에 반하여 첫 번째 치료를 한 후 증상이 다시 악화될 때까지의 기간이 상대적으로 더 길거나 3회 이상 반복되는 증상 악화가 있을 때는 A-CIDP의 가능성이 높을 것으로 판단하였다[36,81].
3) 새로운 치료제
활성화된 보체는 AIDP와 AMAN 모두에서 신경손상을 일으키는 중요한 인자로 알려져 있으며 이를 억제하는 약제들의 임상시험이 진행 중에 있다. Eculizumab은 보체C5억제제(complement C5 inhibitor)로 두 개의 이중맹검무작위대조시험(double-blinded randomized controlled trial) 임상2상이 진행되었다[82,83]. 두 연구 모두 동일한 방식을 사용하였고 각 연구는 두 군으로 나누어 진행하였는데, 한 군은 eculizumab과 면역글로불린정맥치료를, 다른 한 군은 위약(placebo)과 면역글로불린정맥치료를 받았다. 스코틀랜드 그룹의 연구에서는 총 7명(실험군 5명, 대조군 2명)의 적은 환자가 등록되어 임상증상의 호전을 판단하기에 어려움이 있었으나[82], 일본의 연구에서는 35명(실험군 23명, 대조군 12명)의 환자들이 등록되어 eculizumab으로 치료받은 군에서 24주 이내에 달리기가 가능한 환자의 비율이 높은 것을 확인하였다[83]. Eculizumab은 비교적 안전한 약제로 생각되지만 치료받은 2명의 환자에서 패혈증을 비롯한 치명적인 합병증이 보고되어 이에 대한 주의가 필요하며[83], 치료 효과를 검증하기 위하여 보다 많은 환자들을 대상으로 한 추가적인 임상연구가 필요하리라 판단된다. 이 외에 보체C1q에 대한 항체(anti-complement factor 1q)가 AMAN과 MFS의 동물모델에서 효과를 보여 이에 대한 임상1상 연구가 준비 중이다[84].
또 다른 연구 중인 치료제는 고름사슬알균(Streptococcus pyogenes)이 분비하는 효소로서 IgG를 분해하는 작용을 한다. 이는 병리작용을 하는 자가항체를 항원이 결합하는 부위인 Fab와 세포에 결합하는 부위인 Fc로 분해하는 역할을 한다. 현재 유럽에서 임상2상을 계획하고 있다[8].
8. 예후
GBS는 매우 다양한 예후를 보인다. 급성기에 적절한 치료를 받으면 약 80%의 환자가 6개월 이후에 부축 없이 보행이 가능한 정도로 회복하나 3-7%의 환자는 적절한 치료에도 불구하고 질병의 전반기에 다양한 합병증으로 사망하는 것으로 알려져 있다[85]. 대부분은 급성기에 호흡기능장애나 이와 연관된 합병증 또는 부정맥과 같은 자율신경장애로 인하여 사망하지만 일부 환자들은 증상이 호전되어 집중치료실에서 일반 병실로 이동한 뒤에 사망하는 경우도 있어 지속적인 호흡기계 및 심혈관계의 감시가 필요하다. 증상의 호전은 GBS의 발병 후 1년 이내에 이루어지지만 3년 이상의 기간까지도 서서히 회복되기도 하며, 기능적으로 발병 전과 유사한 상태로 회복되더라도 지속적인 통증과 피로를 호소하기도 한다. 40세 이상에서 발병하거나 위장관 감염이 선행한 경우, 증상이 심하게 발생한 경우 그리고 당뇨의 병력이 있는 경우에는 예후가 좋지 않은 것으로 알려져 있다[86,87].
결 론
GBS는 100년 이상의 오랜 기간 동안 정립되어 온 대표적인 염증다발신경병이지만 지금까지도 상당 부분에서 병리기전이 밝혀지지 않았고 치료 방법도 매우 제한적이다. 대부분의 환자에서 장기적인 치료가 필요하고, 사망하거나 심각한 합병증이 발생하는 경우도 적지 않다. 따라서 좋은 예후를 기대하기 위해서는 최대한 이른 시기에 GBS를 진단하고 면역치료를 시작해야 하며 다양한 자율신경기능장애와 합병증에 대하여 대비하고 이에 맞는 적절한 치료를 계획해야 한다. 장기적으로는 면역병리기전에 대한 연구가 더 많이 이루어져서 다양한 표적치료제가 개발되기를 기대한다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print