인간프라이온병(human prion disease)은 정상 프라이온 단백의 구조 변화로 발생한 변형프라이온단백(scrapie isoform of the prion protein)으로 인하여 유발되는 매우 드물고 치명적인 신경퇴행질환이다. 프라이온질환은 빠르게 진행하는 치매와 이상행동, 실조, 추체외로증상으로 발현하여 수개월 내에 사망에 이른다. 인간프라이온병 중에 10-15%를 차지하는 유전프라이온병(genetic prion disease)은 20번 염색체의 프라이온 단백 유전자(prion protein gene, PRNP)의 변이에 의하여 발생한다[1]. 유전프라이온병 중 하나인 게스트만-슈트로이슬러-샤잉커병(Gerstmann-Sträussler-Scheinker, GSS)은 서서히 진행하는 소뇌실조가 특징적이며 질병 후반기(2-4년째)에 인지기능 저하가 동반된다[2]. GSS와 관련된 가장 흔한 돌연변이는 PRNP 102번 코돈의 프롤린(proline)이 류신(leucine)으로 치환된 과오돌연변이(P102L)이다[1]. 저자들은 빠르게 진행하는 인지기능 저하와 뇌 자기공명영상(magnetic resonance imaging, MRI) 소견 그리고 가족력이 없다는 점에서 산발Creutzfeldt-Jakob병(Creutzfeldt-Jakob disease, CJD)을 먼저 의심하였으나 GSS의 가장 흔한 돌연변이인 P102L가 확인된 증례가 있어 이를 보고하고자 한다.
증 례
48세 여성이 3주만에 급격하게 진행하는 인지저하로 응급실에 왔다. 환자는 대졸의 초등학교 교사로, 환자가 한 달 전까지 살았던 집 주소를 기억 못하는 것을 환자의 남편이 방문 3주 전에 처음으로 발견하였다. 외우고 있던 자신의 반 학생들의 이름을 기억하지 못하여 출석부를 보고 학생들의 이름을 부르게 되었고, 간단한 계산을 할 수 없게 되었으며, 자판의 위치를 기억하지 못하여 컴퓨터 업무를 할 수 없게 되었다.
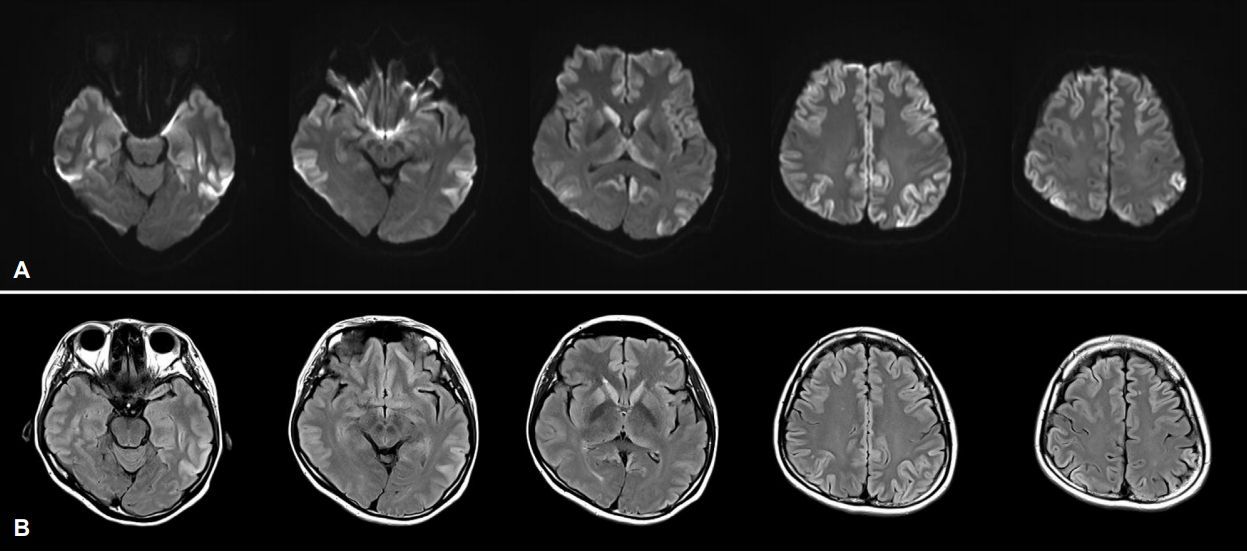
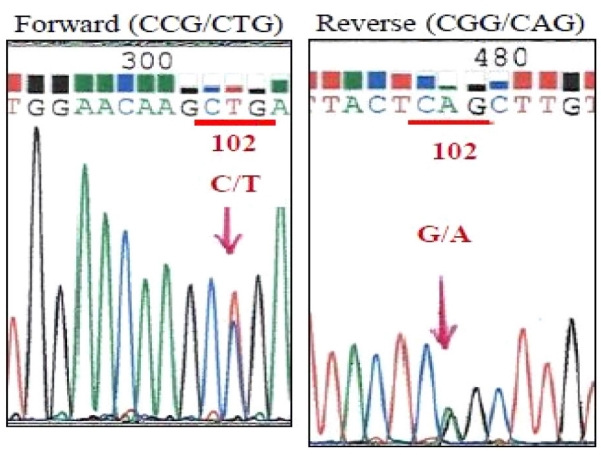
과거력에서 이상은 없었고 신경퇴행질환과 관련된 가족력도 없었다. 응급실 방문 시 신경학적 진찰에서 운동, 감각기능 이상이나 파킨슨증은 없었고, 소뇌기능검사와 보행은 정상이었다. 움켜잡기반사, 손바닥턱끝반사, 입내밀기반사, 눈썹사이반사는 관찰되지 않았고, 안면구강실행증은 없었으나 사지관념운동실행증을 보였다. 증상 발생 3주째에 시행한 한국판간이정신상태검사는 16/30점이었고, 언어 평가시 이해력이나 유창성, 따라 말하기에 이상 소견은 없었으나 볼펜을 제시하였을 때 연필로 말하고, 날씨를 ‘날시’로 적는 맞춤법 오류를 보였다. 증상 발생 8주째에 시행한 한국판간이 정신상태검사는 10/30점으로 더욱 악화되었고, 서울신경심리선별종합검사에서 주의집중력, 기억, 언어, 시공간, 전두엽/집행기능 전영역에서 0.01%tile 이하로 심한 인지기능 저하가 확인되었다(Table 1). 언어의 유창성, 이해력, 따라 말하기, 이름 대기의 모든 부분에서 정상 범주 아래로 악화된 것을 확인할 수 있었고, 간단한 문장을 읽고 이해는 하였으나 쓰기에서는 본인의 이름도 쓰지 못하였다. 사지관념운동실행증은 관찰되었으나 안면구강실행증은 없었다. 전혈구검사, 간기능, 신장기능, 갑상선호르몬, 비타민B12, 매독선별검사를 포함하는 혈액검사에서 이상은 없었고, 뇌파검사도 정상이었다. 확산강조영상(diffusion-weighted imaging, DWI)과 액체감쇠역전회복영상(fluid-attenuated inversion recovery imaging, FLAIR)에서 광범위한 대뇌피질과 기저핵의 고신호강도와 시상베개의 고신호강도(pulvinar sign)가 보였다(Fig. 1). 매우 빠르게 진행하는 치매와 DWI와 FLAIR에서 광범위한 고신호강도를 확인한 후 산발CJD을 감별하기 위하여 뇌척수액검사를 진행하였다. 뇌척수액에서 14-3-3단백과 변형프라이온단백이 검출되었고, 타우단백질도 증가된 소견이었다(10923.8 pg/mL). PRNP의 polymerase chain reaction시퀀싱에서 P102L 이형접합 돌연변이가 발견되었고 유전자 다형태(polymorphism)는 없었다(Fig. 2).
발병 3개월까지는 신경학적 진찰에 협조가 되어 소뇌기능 평가를 할 수 있었는데 정상이었다. 발병 4개월 이후로 심한 인지기능 저하로 인하여 자발적인 발화가 거의 없고 대소변 처리를 스스로 할 수 없게 되었으며, 갑자기 울거나 화를 내는 감정 불안정성도 동반되었다. 발병 8개월 뒤에는 전혀 걷거나 말할 수 없게 되었고, 삼킴곤란이 발생하여 코위관을 삽입하였다. 발병 9개월에 우측 안구편위를 동반한 반복적인 근간대경련이 발생하여 시행한 뇌파검사에서 좌측 두정엽에 예서파복합과 간헐 삼상파(episodes of sharp and slow wave complexes and intermittent triphasic waves)가 확인되었다.
고 찰
2018년도 미국 질병통제예방센터가 채택한 유전프라이온병의 확정 진단은 병리조직 소견과 1차 친족의 가족력 또는 PRNP의 발병 돌연변이가 필요하다. 추정 진단은 진행하는 신경정신행동질환과 1차 친족의 가족력 또는 PRNP의 발병 돌연변이가 확인될 때 진단이 가능하며 P102L은 GSS와 관련된 돌연변이로 제시되고 있다[3]. 우리 환자는 진행하는 신경정신행동질환과 GSS와 관련된 돌연변이가 발견되어 최종적으로 GSS로 추정 진단을 내릴 수 있었다. 유전프라이온병은 임상병리학적 특징에 따라 유전CJD, GSS, 치명적가족불면증(familial fatal insomnia)의 3가지로 분류된다. 이 중 GSS는 특징적인 증상이 진행하는 소뇌실조라고 알려져 있으며 발병 후 2-4년이 경과한 시기에 인지저하가 동반되며, 평균 기대 여명은 5년 정도로 다른 프라이온질환에 비하여 유병 기간이 길다[2]. 산발CJD와 다른 유전프라이온병에 비하여 뇌 MRI나 뇌파에서 이상 소견이 드물고 14-3-3단백 양성률이 낮다[4,5]. 이와 비교해 보았을 때 우리 환자의 경우 초기 증상으로 실조를 포함한 소뇌 증상이 전혀 없었고, 인지저하가 병의 시작부터 매우 빠르게 진행된 점과 초기 뇌 MRI에서의 광범위한 변화와 14-3-3단백 양성은 전형적인 GSS의 표현형과는 달랐다. 반면에 산발CJD 진단기준에 따르면 1) 빠르게 진행하는 인지기능 저하, 2) 근간대경련, 시각 또는 소뇌징후, 추체로/추체외로징후, 무운동무언증 중에 적어도 2가지 이상, 3) 전형적인 뇌파 소견(주기예파), DWI 또는 FLAIR에서 꼬리핵/조가비핵 또는 2군데 이상의 피질(측두엽, 두정엽, 후두엽) 부위 고신호강도, 뇌척수액에서 14-3-3단백 중에 적어도 1가지 이상이 만족되면 추정 진단을 내릴 수 있으며, 또는 진행하는 신경계증후군과 뇌척수액에서 변형프라이온단백이 검출되어도 추정 진단을 내릴 수 있다. 따라서 우리 환자의 전체 경과는 돌연변이만 제외한다면 산발CJD 진단기준에도 부합한다.
이와 같이 P102L돌연변이를 가지더라도 전형적인 GSS의 소뇌 실조 표현형이 아닌 증례가 보고되기 시작하면서 P102L의 표현형 이질성(heterogeneity)이 제기되었다[6]. 심지어 한 가계 내에서도 다양한 첫 증상이 보고된 적이 있으며, 대다수가 보행장애로 시작을 하고 인지기능 저하는 수년 뒤에 생겼지만 인지기능 저하가 첫 증상인 환자도 있었다[6,7]. 일본의 한 연구에서는 P102L돌연변이를 가지는 환자 57명을 임상증상에 따라 2가지로 분류를 하였다[4]. 초기에 소뇌 증상이 나타나는 경우를 GSS표현형이라고 명명하였고 이는 전체 P102L 환자의 79%를 차지하였다. 반면에 초기에 빠르게 진행하는 치매로 발병한 경우를 CJD표현형이라고 하고 이는 21%였다. P102L-GSS와 P102-CJD의 평균 발병 연령은 55세로 유사하나 P102L-CJD가 P102-GSS에 비하여 더 짧은 유병 기간을 보이며, 근간대경련, 무운동무언증이 더 흔히 나타나고, 뇌 MRI에서 고신호강도, 뇌척수액의 타우단백의 증가, 뇌파에서 주기예파(periodic sharp wave complexes)를 더 흔히 동반한다고 보고되었다[4]. 우리 환자는 빠르게 진행하는 치매로 발병을 하였고, 근간대경련, 무운동무언증, 뇌 MRI와 뇌파에서 이상 소견이 모두 확인되어 P102L-CJD표현형과 부합하였다. 국내에서도 P102L를 가진 증례 1명이 보고된 적이 있으나 보행장애와 구음장애로 증상이 시작되었고 발병한지 2년 뒤부터 인지기능 저하가 시작되었다고 기술되어 있어서 전형적인 GSS표현형이었고 비슷한 증상의 가족력이 있었다[8].
우리 환자는 돌연변이가 발견되었지만 관련된 가족력이 전혀 없었다. GSS는 상염색체 우성으로 유전된다고 알려져 있지만 이와 같이 유전프라이온병의 많은 증례 들에서 가족력이 없는 경우가 많아 최근에는 가족성(familial)프라이온병이라는 용어보다 유전(genetic)프라이온병이라는 용어가 선호되기도 한다[1]. 유전프라이온병으로 진단되었으나 가족력이 없는 이유에 대하여 정확한 가족력을 모르거나 선대가 일찍 사망하거나 자식이 없어 확인이 안 된다고 설명을 하기도 하지만 de novo로 발생한 PRNP돌연변이가 확인된 바도 있다[9,10]. de novo P102L은 102번 코돈에서 자발적인 디메틸화로 인하여 사이토신에서 티미딘으로 변화되며 P102L의 치환이 발생하였을 것이라는 가설이 제시되기도 하였다[10].
이 증례는 GSS의 가장 흔한 원인이라고 알려진 P102L돌연변이를 가지고 있으나 GSS표현형이 아닌 CJD표현형으로 발현한 국내 첫 보고이다. P102-CJD표현형은 임상 특징, MRI, 뇌파, 14-3-3단백만으로는 산발CJD, 유전CJD와 구분이 어려워 PRNP 유전 분석이 필요하다. 같은 돌연변이에서 임상 경과가 다른 이유에 대하여 아직 밝혀져 있지 않아 향후 병리조직 연구가 추가적으로 더 필요할 것이다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print