TPM3유전자 이형접합 과오돌연변이에 의한 캡근병증
Cap Myopathy With a Heterozygous TPM3 Missense Mutation
Article information
Trans Abstract
Cap myopathy is pathologically characterized by cap structures comprising well-demarcated areas under the sarcolemma and containing deranged myofibrils and scattered Z-disks. Clinically it presents with slowly progressive muscle weakness, myopathic face, and frequent respiratory insufficiency. Four genes have been reported to be associated with the disease: TPM2, TPM3, ACTA1, and NEB. Here we describe that a patient presenting with mild limb weakness with facial affection showed cap structures on muscle pathology and carried a heterozygous TPM3 mutation.
캡근병증은 매우 드문 선천근병증의 아형으로 병리조직학적으로 골격근 섬유 내부에 모자(‘cap’) 형태의 비정상적인 구조물이 관찰된다[1]. 임상적으로는 주로 유소아기에 발병하여 서서히 진행하는 근력약화와 근긴장도의 저하가 특징이다. 사지 전반에 비슷한 정도의 위약이 생기며 이와 더불어 안면부 근위약, 높은 구개, 외안근마비와 호흡근의 침범이 나타날 수 있다[2]. 캡근병증의 원인은 2007년 TPM2 (β-tropomyosin)유전자에서 이형접합결손을 처음으로 보고한 이후[3] TPM3 (α-tropomyosin), ACTA1 (α-actin) 그리고 최근에는 NEB (nebulin) 유전자에서도 돌연변이를 보고하였다[4-6].
저자들은 10세 무렵부터 잦은 호흡기계 감염과 사지의 근력저하가 발생하여 근생검을 통해 캡 구조를 확인하고 유전자검사에 의해 TPM3 유전자 돌연변이를 발견하여 캡근병증으로 진단한 증례를 보고한다.
증 례
19세 남자가 5년 전부터 외사시로 안과에서 진료받던 중 안면근위약과 사지 근위약이 관찰되어 신경과로 의뢰되었다. 10세 이후부터 체중이 증가하지 않아 점차 마른 체격으로 변하였고 운동능력이 저하되어 달리기에서 항상 꼴찌를 했다고 한다. 비슷한 시기에 내반족 변형으로 인한 교정술을 받았으며, 16세에는 척추 측만증 교정 수술을 받았다고 한다. 또한, 호흡기계 감염이 매우 잦았으며, 중환자실에서 인공호흡 보조를 통한 치료를 두 차례 받기도 했다. 출생 당시와 주산기에 문제는 없었고 영유아기에 정신운동 발달 역시 정상이었으며, 10세 이전에는 또래와 비교해 체격이 좋은 편이었고 운동능력도 매우 좋았다고 한다. 아버지는 심근경색이 있었으며 어머니와 여동생은 건강하였다.
신경계 진찰에서 매우 마른 체형이었고 근육병증 얼굴과 양안의 눈꺼풀처짐이 있었다. 안구운동검사에서는 모든 방향에서 경미한 외안근 마비가 있었고, 높은 구개를 확인할 수 있었다. 날개어깨뼈(winged scapula)가 양측에 있었다. 도수근력검사에서 경부굴근이 Medical Research Council (MRC) 3등급, 양측 상지 몸쪽 근육은 4등급, 아래팔근육은 4-~5-등급으로 측정되었으며 수부 근력은 비교적 잘 유지되어 있었다. 양측 하지 몸쪽 근육은 5등급, 먼쪽 근육은 4등급이었다. 심부건반사는 사지에서 모두 소실되었으며 병적과다 반사는 나타나지 않았다.
혈청 크레아틴키나아제는 119 IU/L (정상치, 5-217 IU/L)로 정상이었다. 신경전도검사는 정상이었으며 침근전도검사에서는 우측 공통손가락폄근(extensor digitorum communis), 우측 위팔두갈래근(biceps brachii)과 우측 앞정강근(tibialis antrior)에서 자발 전위는 뚜렷하지 않았으나 진폭이 낮고 지속시간이 짧은 다상운동단위전위를 보여 근병증에 합당한 것으로 판단했다. 근육 컴퓨터단층촬영에서 위팔, 아래팔과 넓적다리에서는 이상이 없었으나 양측 하지의 앞정강근과 장딴지근에서 경도의 근 위축과 지방화를 확인하였다.
좌측 앞정강근 근생검에서는 근세포 크기의 다양성이 증가되었으며 근세포는 전반적으로 둥근 모양이었다. 특히 일부 근세포의 가장자리에, 헤마톡실린-에오신 염색에서 호산성, 그리고 변형 고모리-트리크롬 염색에서는 보라빛으로 염색되는 캡 구조가 보였다(Fig. 1A, B). ATPase 염색에서도 캡 구조는 상대적으로 옅게 염색되어 구별되었으며, 1형 근섬유가 수적으로 우세했다(Fig. 1C). 전자현미경에서 캡 구조의 내부는 근원섬유 구조가 파괴된 상태에서 A-밴드가 소실되고 Z-disk와 유사한 전자 밀도를 보이는 다수의 물질이 흩어져 있었으며 사립체가 결핍되어 있었다(Fig. 1D). 캡 구조의 경계는 동일 근 세포 내에서 근원섬유가 정상적으로 배열된 영역과 잘 구분할 수 있었다.
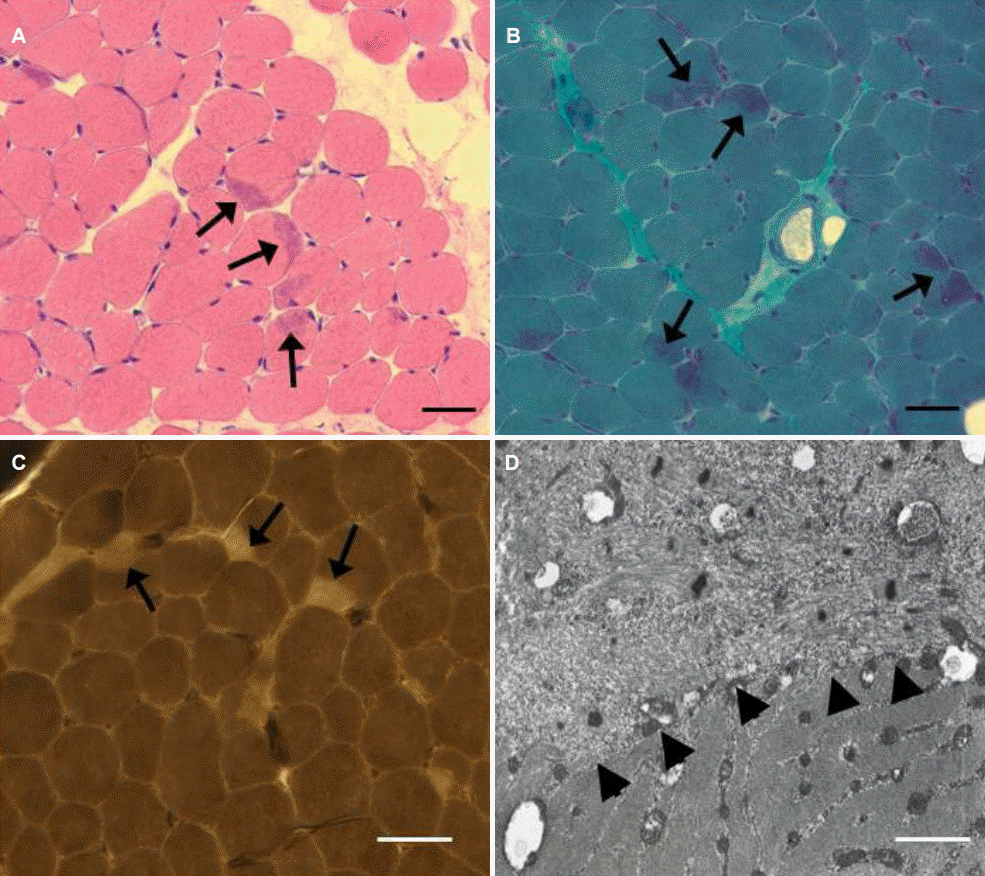
Muscle pathology. (A, B) Increased fiber size variation and round-shaped muscle fibers were seen, and crescent-shaped structures (cap structures) were occasionally found in the periphery of muscle fibers (arrows). (C) Myofibrillar ATPase staining at pH 4.3 shows type 1 fiber predominance and pale-stained cap structures (arrows). (D) On electron microscope, myofibrillar arrangement was disorganized and electron-dense materials similar to Z-disk were scattered (arrowheads). (A, H&E stain; B, modified Gomori-Trichrome stain; C, ATPase pH 4.3; Bar = 50 μm in A, B and C; 1 μm in D).
근생검을 바탕으로 캡근병증으로 진단하였고 원인 유전자로 알려져 있는 TPM2, TPM3, ACTA1과 NEB유전자의 돌연변이를 검색하기로 했다. TPM3유전자에 대한 염기서열분석에서 이형접합과오돌연변이인 p.R168C (c.502C>T, reference sequence NM_15 2263.2)를 확인할 수 있었다(Fig. 2). 이는 동일 질환에서 이미 보고한 적이 있으므로[4] 이 환자도 이 질환과 연관한 원인 돌연변이일 것으로 생각하고 다른 유전자검사는 더 시행하지 않았다. 부모의 유전자검사를 권유하였으나 환자가 거부하였다. TPM3유전자와 연관한 캡근병증의 경우 모두 이형접합체로 보고하였으며 환자의 부모는 유사 증상이 나타나지 않았기 때문에 환자의 돌연변이는 de novo일 것으로 판단하였다.
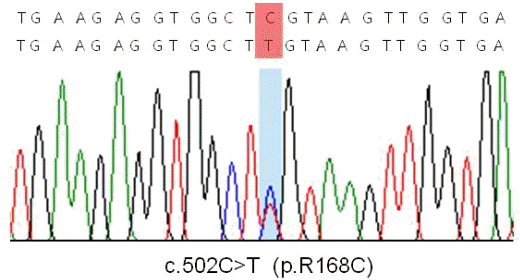
Chromatogram of TPM3 sequences. A heterozygous missense mutation of p.R168C (c.502C>T) by reference sequence NM_152263.2 is seen.
고 찰
캡근병증은 매우 드문 질환으로 전형적인 선천근병증의 임상 양상을 보이면서 골격근 병리에서 캡 구조라는 특징이 나타난다[1]. 캡 구조는 광학 현미경에서 근 세포의 바깥쪽에 구별되는 염색 영역을 말하는데, 본 증례와 같이 전자현미경에서 근원섬유 구조가 완전히 파괴되고 확대된 Z-disk가 산재되어 있는 형태로 나타나는 것이 일반적이다[3]. 현재까지 이 질환과 연관하여 알려져 있는 원인 유전자(TPM2, TPM3, ACTA1, NEB)는 모두 근세포 수축 장치(contractile apparatus)를 구성하는 단백질을 암호화한다. 이들 모두가 네말린근병증의 원인 유전자와 중복되고, 미세구조에서 Z-disk의 이상이 공통적으로 나타난다는 점은 두 질환이 발병기전을 공유할 것임을 시사한다. 심지어 동일한 돌연변이에서 두 질환이 모두 발생되는 것으로 보고하여 유전형이 아닌, 알려지지 않은 어떤 원인에 의해 서로 다른 병리가 발생하는 것으로 생각한다[6]. 두 질환은 임상적으로는 잘 구분되지 않을 뿐만 아니라, 최근에는 한 환자에서 캡 구조와 네말린 소체가 함께 나타난 증례를 보고하여 향후 두 질환의 연관성에 대한 연구가 필요하다[6,7].
본 환자에서 캡 구조는 하나의 근세포다발(fascicle) 중 4-5개의 근 세포에서 나타났는데(~10%) 일부의 보고에서는 캡 구조의 빈도와 임상예후를 연관시키기도 했다[2]. 즉, 근조직 소견에서 캡 구 조가 많을수록 어린 나이에 발병하며, 70% 이상의 근세포에서 캡 구조를 보인 경우 더욱 치명적인 경과를 가진다는 것이다[2]. 본 증례에서 비교적 경한 근력 저하와 안정적인 임상 경과는 캡 구조의 빈도가 낮기 때문으로 추정할 수 있다. 환자는 운동능력의 저하가 나타나기 전인 10세까지 운동 발달에는 문제가 없었고 이후로도 근력 약화로 인한 불편감을 크게 느끼지 못했다. 신경계 진찰을 통해 사지 근력 약화, 눈꺼풀처짐, 외안근마비와 높은 구개를 확인하고 나서야 선천근병증을 의심할 수 있었다. 이 외에 잦은 호흡기계 감염의 병력과 비교적 어린 나이에 두 차례의 인공호흡 보조를 받은 경험이 있었는데, 이러한 호흡기능 저하와 잇따른 호흡부전은 캡 근병증의 특징이다. 지금까지 보고한 거의 모든 환자에서 폐활량이 감소되고 많은 수의 환자가 침습 또는 비침습 호흡보조를 받은 것으로 미루어[3,5,6,8,9] 호흡 관리는 이 질환의 환자에서 가장 중요한 부분으로 생각한다.
TPM3는 캡근병증과 네말린근병증 이외에도 선천근병증 중 하나인 선천섬유형불균형(congenital fiber type disproportion)의 원인유전자로도 알려져 있다. 이 유전자는 slow type의 알파-트로포미오신(α-tropomyosin)을 암호화하며, 이는 주로 1형 근섬유에서 액틴과 결합하여 액틴과 미오신의 상호 작용을 매개함으로써 골격근 수축-이완에 기여한다. 캡 구조의 발생기전과 관련한 보고에서 2차원 전기영동(2D-electrophoresis)을 통해 돌연변이에 의한 알파-트로포미오신 단백질의 분자량과 전하량의 변화가 캡 구조의 형성에 기여할 가능성을 제시하였다[8.9]. 본 증례에서도 이를 근거로 2차원 전기영동을 실시하였으나 정상 단백질과 비교하여 알파-트로포미오신 단백질의 분자량과 전하량 변화는 없었다(data not shown). 따라서, 캡 구조의 형성에는 단백질의 정성 또는 정량 변화 이외에도 다른 기전이 작용할 가능성이 높다. 한 예로, Arg168이 액틴 결합위치에 매우 인접해 있으므로 본 증례와 같이 이 위치에 발생한 돌연변이는 액틴과의 친화력이 현저하게 감소하여 근력저하가 나타난다는 기전이 제시된 적이 있다[6].
결론적으로, 본 증례는 사지의 근력저하와 안면근과 외안근마비, 잦은 호흡기계감염을 보였고 근 생검을 통해 캡 구조를 확인하고 유전자 검사를 통해 TPM3유전자 돌연변이를 확인함으로써 캡근병증을 확진하였다. 캡근병증은 임상적으로 다른 선천근병증과 매우 유사하므로 임상 양상을 통한 감별은 불가능하다. 그러나, 근병리에서 특징적인 캡 구조를 확인한다면 쉽게 유전자 진단까지 할 수 있는 질환으로 생각한다. 캡근병증은 매우 드문 질환으로 임상의에게 선천근병증 진단에 도움이 될 것으로 판단하여 본 증례를 보고한다.
Acknowledgements
본 연구는 2015년도 부산대학교병원 임상연구비 지원으로 이루어졌음.