유전강직하반신마비 환자에서 발견된 SPAST유전자의 Arg450Cys 돌연변이
Arg460Cys Mutation in SPAST Gene in Patients with Hereditary Spastic Paraplegia
Article information
Trans Abstract
Hereditary spastic paraplegia is a group of genetically and clinically heterogeneous neurodegenerative disorder characterized by progressive lower extremity weakness and spasticity. A 49-year-old man presented with progressive gait disturbance. Neurological examination showed spasticity, hyperreflexia in lower extremity and bilateral ankle clonus. He had a family history consistent with autosomal dominant inheritance. Gene testing revealed a heterozygous missense mutation (c.1378C>T [p.Arg460Cys]) in SPAST gene. We report a first Korean family with Arg460Cys mutation in SPAST gene.
유전강직하반신마비(hereditary spastic paraplegia)는 임상 및 유전학적으로 이질적인 질환으로 하지강직과 위약이 특징이다. 임상적으로 순수형과 합병형으로 구분하고, 하지강직과 위약 이외에 치매, 정신지체, 뇌전증, 피라미드외로운동장애, 실조, 난청, 말초신경병, 시신경병 등이 동반되면 합병형으로 분류한다. 보통염색체우성, 보통염색체열성, X 연관 양식으로 유전되며 보통염색체우성의 순수형이 가장 흔한 유형이다[1]. 유전강직하반신마비와 관련된 가장 흔한 유전자 이상은 SPAST 유전자의 돌연변이지만 유전형과 표현형의 연관성이 명확하지 않고, 돌연변이의 민감점(hot spot)이 알려져 있지 않아 진단을 위해서는 SPAST 유전자의 유전부호 전체를 확인해야 한다[2]. 최근 한국 내연구에 따르면 보통염색체우성유전을 보이는 유전강직하반신마비 환자의 66%에서 SPAST 유전자 돌연변이가 확인되었고, 이는 이전 국내연구와 비슷한 결과를 보였다[3,4]. 저자들은 진행하는 하지강직과 위약을 보이며 보통염색체우성 유전 양상의 가족에서 국내에서 보고되지 않은 SPAST유전자의 돌연변이를 확인하여 보고한다.
증 례
49세 남자(II-3, Fig. 1)가 10년 전부터 점차 진행하는 보행장애로 내원하였다. 양하지가 무겁고 뻣뻣하여 걸음걸이가 불안정하고, 특히 계단을 내려올 때가 가장 힘들다고 하였다. 과거력에서 특이병력은 없었다. 신경학적진찰에서 의식상태, 뇌신경 기능, 근력과 감각기능은 정상이었다. 심부건반사는 양쪽무릎과 발목에서 항진되었고 발목간대가 나타났다. 소뇌기능은 정상이었고 환자는 강직보행을 보였다. 가족력에서 환자의 어머니와 19세인 둘째 아들이 보행장애를 갖고 있었다. 환자의 어머니(I-2, Fig. 1)는 50세 경부터 걷기 어려운 증상이 발생해 이후 점차 진행하여 집에서만 생활하다가 68세에 뇌출혈로 사망하였다. 환자의 아들(III-5, Fig. 1)은 17세경부터 계단을 내려올 때 불편한 증상이 시작되었고, 신경학적 진찰에서 근력은 정상이나 무릎반사와 발목반사의 항진 및 바빈스키징후가 관찰되었다. 환자의 가족력을 고려하면 보통염색체우성유전 양상으로 판단하였다(Fig. 1).
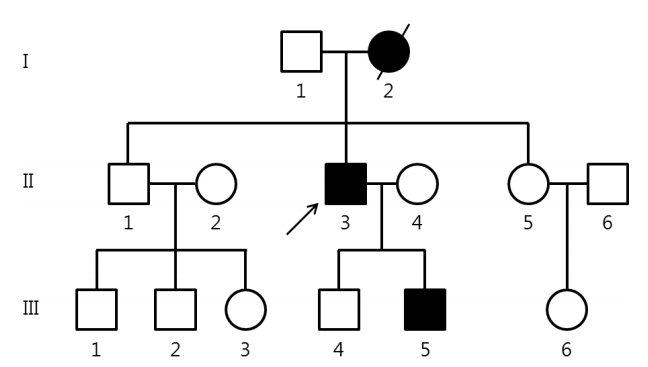
The pedigree of a patient. The pedigree of a family demonstrated an autosomal dominant inheritance. Arrow indicates index patient.
간이정신상태검사와 뇌, 척수자기공명영상, 갑상선기능, 비타민B12, 매독, 구리(copper), 사람T세포림프친화바이러스(HTLV), 사람면역결핍바이러스(HIV), 자가면역항체, 뇌척수액검사는 모두 정상이었다. 말초신경계 침범을 확인하기 위해 시행한 신경전도와 근전도검사는 정상이었다. 보통염색체우성을 보이는 순수형의 유전강직하반신마비로 추정되어 SPAST유전자검사를 시행하였다. 환자의 백혈구에서 추출한 DNA를 중합효소연쇄반응(polymerase chain reaction)으로 증폭하여 SPAST유전자의 17개 엑손 및 엑손-인트론(intron) 경계부위의 염기서열을 직접염기서열결정(direct sequencing)방법으로 분석하였다. 분석결과 SPAST유전자의 엑손11에서 c.1378C>T (p.Arg460Cys) 이형접합돌연변이가 발견되었다(Fig. 2). 환자의 아들에서도 같은 과오돌연변이가 확인되었다.

The results of SPAST gene mutation analysis in index patient (II-3). All coding exons and flanking introns of the SPAST gene were amplified by polymerase chain reaction using primers. Direct sequencing analysis demonstrated a heterozygous C to T substitution in exon 11 of the SPAST gene (c.1378C>T), which caused an Arg460Cys missense mutation (arrow). The affected amino acid is located in the AAA cassette region of the protein. The arrow shows the position of a C-to-T transition at nucleotide 1378. AAA; ATPase associated with diverse celluar activity.
고 찰
유전강직하반신마비는 신경퇴행질환으로 하행피질척수로와 상행척수로말단부의 축삭변성이 발생하며, 이 결과로 양쪽 다리에 진행하는 강직과 위약이 나타나고 진동감각소실이나 배변장애가 동반될 수 있다. 현재까지 다양한 유전양식의 많은 돌연변이가 보고되었고, 이 중 SPAST유전자의 돌연변이가 가장 흔하다. SPAST유전자돌연변이는 순수형보통염색체우성유전을 보이는 환자의 15-40%, 산발성 환자의 10%에 해당한다[5]. 본 증례도 양쪽 다리의 강직과 심부건반사항진 이외 다른 신경계증상을 보이지 않았고, 가족력을 고려하면 순수형의 보통염색체우성유전일 가능성이 높아 SPAST유전자검사를 우선 시행하였다. 유전강직하반신마비의 한 유형인 강직하반신마비4형(spastic paraplegia 4, SPG4)은 염색체 2번에 위치한 SPAST유전자의 돌연변이로 발생하며, 이 유전자는 ATPase associated with diverse celluar activity (AAA) ATPase의 일종인 spastin을 부호화한다. spastin은 AAA를 통해 미세관(microtubule)에 작용하여 축삭성장과 분지에 관여하는 것으로 알려져 있다[6]. SPG4는 오랫동안 다른 신경계증상을 동반하지 않는 순수형으로 간주되었지만, 진단된 환자의 50%에서 감각이상, 괄약근장애, 다발신경병을 동반할 수 있다. 또한 양쪽 팔의 강직, 실조, 인지장애 등이 나타난다. 최근 연구는 SPG4 환자에서 인지장애와 치매를 주요한 표현형으로 포함하고 있으며, 양성자자기공명분광법이 인지장애진단에 도움이 된다[7]. 국내 유전강직하반신마비 환자에 대한 유전자분석 연구에서 보통염색체우성유전 환자의 64-66%에서 SPAST유전자돌연변이로 확인되어 외국연구보다 높은 비율을 보였다. 임상적으로 일부 환자에서 배뇨장애나 진동감각 저하를 보였지만 대부분의 환자는 하지 강직과 위약이 주증상인 순수형으로 분류되었다[3,4].
SPAST유전자 이상은 주로 무의미돌연변이, 과오돌연변이, 절단돌연변이로 알려져 있다. 발병연령이나 임상중증도와 관련하여 돌연변이 유형에 따른 표현형의 유의미한 차이는 없지만, 메타분석에서 과오돌연변이 환자가 다른 돌연변이에 비해 발병연령이 빠른 경향을 보였다[1]. 하지만 같은 돌연변이를 갖는 가족 내에서도 발병연령이나 중증도는 많은 변이를 보인다. 본 증례에서 확인된 c.1378C>T (p.Arg460Cys)돌연변이는 과오돌연변이로 2004년에 처음 보고되었고[8], 주로 순수형의 보통염색체우성유전에서 확인되고 있다. 하지만 증례와 같은 유전자이상을 보인 환자들의 임상양상은 많은 변이를 보였다. 처음 보고된 환자는 본 증례와 달리 상지 침범이 뚜렷하였고, 다른 환자는 발병연령이 10세 이전으로 보고되었다[2,8]. 가족력이 없는 산발성 환자의 10%에서 SPAST유전자 돌연변이가 확인되고, 합병형보다 순수형에서 발생빈도가 높다. 산발성 환자에서도 본 증례와 발병연령이나 임상양상이 유사하고, 동일한 유전자이상이 확인된 환자가 보고되었다[9]. 또한 유전자검사방법의 발달로 SPAST유전자에서 결손돌연변이를 포함한 새로운 돌연변이가 보고되고 있어 유전형의 이질성이 SPG4의 특성으로 이해되고 있다.
보통염색체우성유전을 보이고 다른 신경계증상을 동반하지 않는 순수형은 SPG4 이외에 SPG3, SPG6, SPG10, SPG12, SPG13, SPG19, SPG31, SPG37, SPG40, SPG41, SPG42유형을 고려해야 한다[1]. 이 중 SPG3A, SPG10, SPG31을 제외하고 임상적으로 SPG4와 감별이 어렵다. SPG3A는 보통염색체우성유전에서 SPG4 다음으로 흔하지만 대부분의 환자가 10세 이전에 증상이 시작되어 SPG4와 차이를 보인다. 하지만 국내연구에서는 보통염색체우성유전을 보인 환자들에서 SPG3A 유전자돌연변이 발생이 외국에 비해 낮은 것으로 보고되었다[3]. 이러한 차이는 국내연구대상 환자의 수가 적고, 한국인과 외국인의 인종간 차이에 기인한 것으로 사료된다. SPG10, SPG31 환자는 말초신경병과 Silver 증후군을 동반하는 것이 특징이다[10]. 임상적으로 구분이 어려운 순수형 유전강직하반신마비에서 보통염색체우성유전의 가족력이 있거나 산발성 환자는 먼저 SPAST 유전자검사를 시행하여 SPG4유형을 확인하는 것이 권장된다. 저자들은 보통염색체우성유전을 보이는 순수형 유전강직하반신마비 가족에서, 아직 국내에서 보고되지 않은 SPAST유전자의 과오돌연변이를 확인하였다.