CHRNA1 유전자변이와 연관된 선천근무력증후군
Congenital Myasthenic Syndrome Related to CHRNA1 Variant
Article information
Trans Abstract
Congenital myasthenic syndromes are a genetically and clinically heterogeneous group of neuromuscular disorders linked by abnormal signal transmission at the motor endplate caused by various genetic defects. Major clinical symptoms include weakness and fatigue during the first years of life but patients may also present with hypotonia, facial weakness, swallowing difficulties, respiratory dysfunction, ptosis and ophthalmoparesis. Here we report a 10-year-old boy who presented with mild developmental delay and bilateral ptosis caused by a frameshift mutation in the CHRNA1 gene that co-segregated within the family, and finally diagnosed as autosomal dominant congenital myasthenic syndrome.
선천근무력증후군(congenital myasthenic syndrome)은 유전신경근육장애로 신경근육접합부(neuromuscluar junction)에 여러 형태의 결함으로 발생하게 된다. 임상적으로는 근긴장저하부터, 근무력, 가벼운 눈 처짐 증상까지 다양하며 일반적으로 다양한 항체검사에서 음성 결과가 나오지만 임상적으로 눈 처짐 및 근력약화가 의심되는 경우 감별진단이 필요한 질환이다. 현재까지 다양한 유전원인이 밝혀져 있으며 국내 최초로 CHRNA1 유전자 이상에 의해 발생한 선천근무력증후군과 이와 동반된 증상들을 보고한다.
증 례
10세 남아가 눈 처짐으로 병원에 왔다. 산전진찰에서 이상은 없었고, 임신 38주에 분만 중 손상 없이 정상적으로 질식분만을 통해 3.1 kg으로 태어났다. 하지만 성장과정에서 또래보다 체격이 왜소하였으며, 생후 16개월에 걷기 시작하는 등의 발달 지연이 확인되었다. 또한 1세경부터 사물을 볼 때 비스듬히 옆으로 바라보고, 초점이 잘 맞지 않아 안과에서 정밀검사 후 경미한 외안근마비가 있다고 들었다. 4세경부터 눈 처짐 증상이 저명하게 나타나기 시작하였다. 관절구축 및 척추이상은 없었으나 또래 평균 키에서 하위 25%로 저신장을 보여 소아청소년과를 방문하였고 당시 골연령이 지연되어 3세로 확인되었다. 7세경에 눈 처짐이 악화되어 안과에서 양측 안검하수 수술을 받았으나 8세경에 다시 눈 처짐이 재발하여, 눈 처짐에 대한 정밀한 검사를 위해 신경과로 의뢰되었다. 신체진찰에서 양안의 눈 처짐과 안검열의 폭이 11 mm/7 mm로 관찰되었으나, 복시 및 뚜렷한 안구운동장애는 확인되지 않았으며 눈 처짐은 일중 변동을 보이지 않았다(Fig. 1). 신경계진찰에서 눈처짐 외에는 이상이 관찰되지 않았고, 중증근무력증 종합척도점수(Myasthenia Gravis Composite Scale)는 2점으로 확인되었다. 항아세틸콜린수용체항체 및 근육특이 타이로신인사화효소(muscle-specific tyrosine kinase, MuSK)에 대한 항체검사에서 모두 정상 범위로 확인되었으며, 전해질검사와 간기능검사, 갑상샘기능검사, 근육효소검사, 류마티스관절염이나 전신홍반루푸스 등의 자가면역질환에 대한 검사 또한 모두 정상이었다. 흉부 컴퓨터단층촬영 및 전신 자기공명영상은 정상이었다. 반복신경 자극검사 또한 정상이었고 피리도스티그민(pyridostigmine) 정맥주사검사에서 뚜렷한 개선은 보이지 않았다. 가족력에서 친할아버지와 아버지가 어릴 때 성장이 조금 늦었고 눈 처짐이 있었다고 하여, 차세대염기서열분석법을 통해 유전자검사를 진행하였다. 이를 통해 CHRNA1 유전자에서 c.783del (p.Lys262ArgfsTer2)변이가 눈 처짐 증상이 있는 환아와 아버지에서 확인되었다(Fig. 2). 이는 American Medical College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) 지침에 따라 발병 변이로 분류되었고, CHRNA1 유전자 결손에 의한 선천근무력증후군으로 진단하였다. 이후 경험적 치료에 근거하여 피리도스티그민 60 mg으로 경구 투여를 시작하였으며, 현재 눈 처짐 증상은 약간 호전되었다.
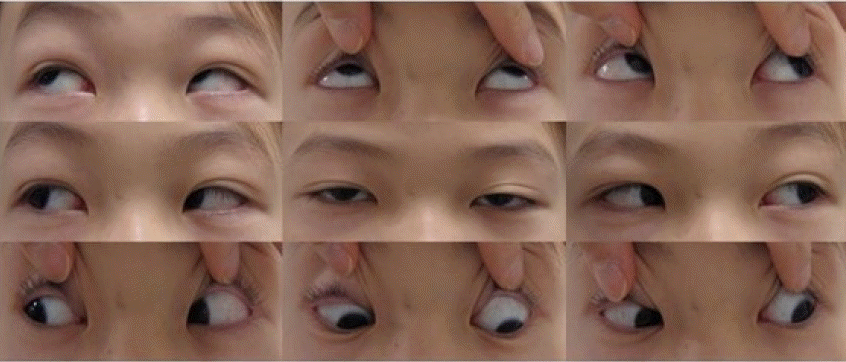
Nine-gaze photography of the proband shows bilateral ptosis without opthalmoplegia.
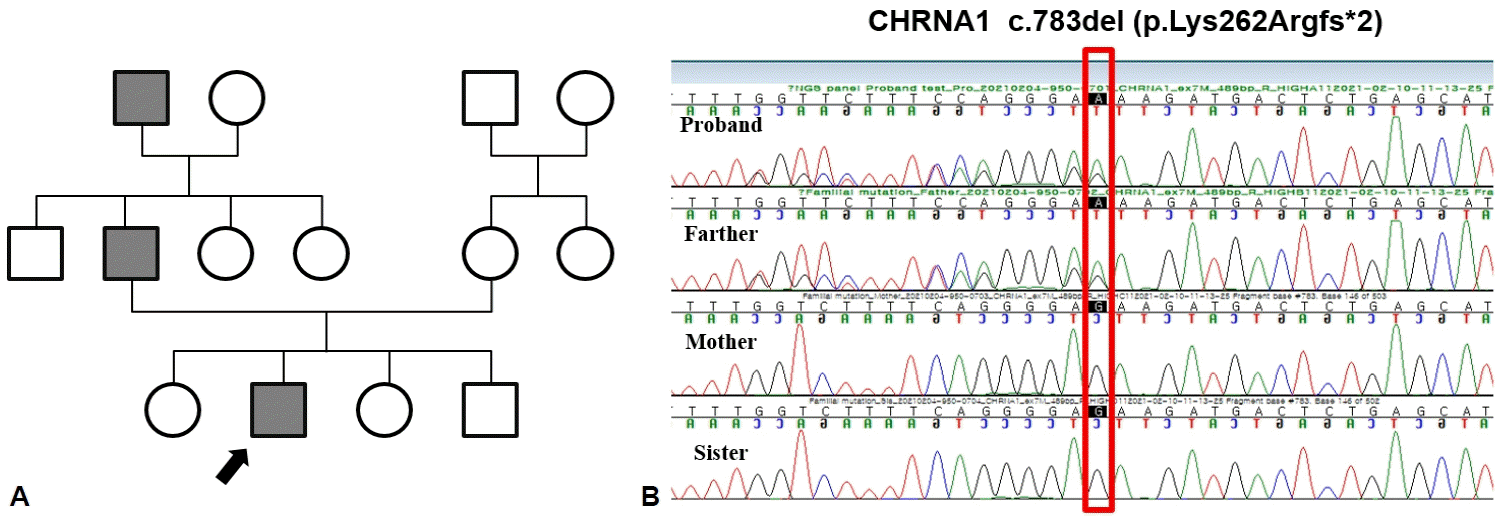
The pedigree of the patient and the family (A). The Sanger sequencing shows the familial segregation with the mutation in the c.783del (p.Lys262fsTer2) of the CHRNA1 gene (B).
고 찰
선천근무력증후군은 신경근육접합부를 구성하고 유지하며 기능하는 데에 필요한 단백질을 형성하는 유전자들의 이상에 의해 발생한다. 선천근무력증후군은 임상적으로 눈 처짐, 운동 시 발생하는 피로, 일시적이거나 지속되는 근력의 약화와 함께 얼굴과 숨뇌 기능의 약화도 동반된다. 하지만 증상 발생 나이나 임상 증상 그리고 치료에 대한 반응이 유전적 결손의 종류에 따라 다르다는 것이 특징이다. 현재까지 약 32개의 원인 유전자가 발견되었으며 아세틸콜린수용체의 유전자 결손뿐만이 아니라 신경근육접합부의 시냅스 이전과 이후에 관여하는 유전자들도 밝혀진 바 있다[1]. 가장 흔한 원인 유전자로는 아세틸콜린수용체의 유전자 결손이 가장 흔하나 단일 유전자 결손으로는 RAPSN, COLQ, DOK7 등이 있다[2]. 아세틸콜린수용체의 유전자 결손이 있는 경우 크게 운동기능(kinetic function)과 아세틸콜린수용체의 발현이상의 두 개의 주된 기능이상으로 나뉜다[2]. 아세틸콜린수용체는 총 5개의 막전위 소단위체가 있으며, 2개의 α, 각각 한 개의 β, δ가 있으며 태아에서는 γ이 존재하는데 태어나면서 ε으로 바뀌는 소단위체로 구성되어 있다. 아세틸콜린수용체의 유전자 결손이 있는 경우 현재까지 가장 흔한 원인은 ε 소단위를 형성하는 CHRNE로 알려져 있다[3]. 하지만 본 증례는 이 중 α 소단위체를 형성하는데 기여하는 CHRNA1의 유전자변이로 최근에 밝혀진 유전자이다. 해당 유전자는 α 소단위체를 형성하는 데 중요한 역할을 하는 유전자이며, 교대잘라이음(alternative splicing)을 통해 엑손 P3A가 존재하는 mRNA와 엑손 P3A가 존재하지 않는 mRNA를 형성하며, 엑손 P3A가 존재하지 않는 mRNA가 기능하는 아세틸콜린 α 소단위체를 형성하는 것으로 알려져 있다[3].
원인 유전자에 따라 임상 양상이 달라지는데, COLQ 유전자와 연관된 선천근무력증후군은 경미한 근쇠약부터 호흡장애까지 다양한 임상 증상을 보일 수 있으나 일반적으로 눈근육은 침범하지 않지만 몸통근육을 심하게 침범하며 척추측만증까지 생기는 경우가 많이 알려져 있다. 하지만 눈 처짐, 눈근육마비 및 안면근육의 약화만을 보인 사례가 보고된 바 있다[1]. 그 다음으로 흔한 DOK7 유전자와 연관된 선천근무력증후군은 보행장애와 함께 근위부 근육운동기능을 침범하여 근육질환과의 감별이 필요한 경우가 많은 것으로 알려져 있다[4]. DOK7 유전자 연관 선천근무력증후군은 눈 처짐은 흔하게 동반되나 눈근육마비는 흔하지 않으며 피곤함이 두드러지게 나타난다고 한다. DOK7 유전자는 MuSK와 결합하여 인산타이로신 인산화효소의 하류 신호체계에 연관되어 있으며, 이로 인해 흥미롭게도 아세틸콜린분해효소 억제제에 효과가 없으며 때로는 증상의 악화를 야기할 수 있어 주의를 요한다. DOK7 유전자 결손이 있는 경우 살부타몰(salbutamol)과 같은 베타2작용제가 더 효과적인 약물로 알려져 있다[1]. 반면 아세틸콜린수용체에서 가장 많은 문제를 차지하는 CHRNE 유전자 결손은 ε 소단위체의 기능 이상을 불러일으킨다. 이 또한 다양한 임상 증상을 나타내는 것으로 알려져 있으며 호흡부전까지 있는 경우도 있으나 대부분 경미한 숨뇌증상과 함께 눈 처짐과 외안근마비가 동반된다. 이 경우 일반적으로는 아세틸콜린분해효소 억제제에 효과가 있으나 종종 악화가 된 보고도 있으며 베타2작용제나 플루옥세틴과 같은 약물에 좋은 반응을 보이는 경우도 있다[1]. 반면 본 증례에서 확인된 α 소단위체 구성을 담당하는 CHRNA1 유전자는 2008년도에 처음 밝혀졌다[1]. 이 보고에서는 동형 접합체의 CHRNA1 유전자 결손이 확인되었으며 눈 처짐, 안면근육마비 및 쉰목소리가 특징이었으나 발달장애는 보이지 않았다. 3세경 근력의 저하가 나타났으나 아세틸콜린분해효소억제제에 근력약화는 호전되었으며 눈 증상은 호전이 없었다고 하였다[5]. 다른 한 증례에서는 이형 접합체의 CHRNA1의 c.896T>G (p.Val269Gly)변이가 확인된 환자를 보고하였다. 이 환자는 눈 처짐과 함께 부분적인 외안근마비와 함께 원위부근력의 약화가 있었다[5]. 본 증례에서는 위에서 언급된 동일한 M2 영역에 위치한 c.783del (p.Lys262ArgfsTer2)에서 확인되었으며 위의 증례와 유사하게 눈 처짐이 경미하게 동반된 경우였다. 더 나아가 환아의 아버지도 눈 처짐이 있어 안검하수교정수술을 이미 받은 상태였다.
선천근무력증후군은 후천중증근무력증과 임상적으로도 차이를 보인다. 후천중증근무력증의 경우는 신생아 일과성 중증근무력증을 제외하면, 만 2세 미만에서는 나타나지 않는 것으로 알려져 있는 반면에 선천성의 경우는 태어날 때부터 증상을 보일 수 있다. 또한 선천성의 경우에는 길어져 있는 얼굴 모양, 턱나옴증(prognathism), 높은입천장(high-arched palate)과 같이 특징적인 얼굴 형태를 보이는 경우가 많다. 또한 후천성의 경우에는 흉선종 및 다른 자가면역질환과 연관이 있는 경우가 많아, 면역억제제에 반응을 보이는 경우가 많은 반면, 선천성 중증근무력증에서는 면역억제 치료의 효과가 미미한 것으로 알려져 있다[6].
증례를 통해 CHRNA1의 유전자이상이 있는 경미한 선천근무력증후군을 경험하였고, 선천근무력증후군에서 다양한 임상 증상이 생길 수 있음을 알 수 있었다. 특히 가족력이 있는 눈 처짐 등의 증상을 가졌으나, 흉선종 혹은 뚜렷한 자가면역성 질환이 확인되지 않고, 대증적인 중증근무력증 치료에 효과가 미미하거나 없는 경우 선천근무력증후군에 대한 유전자검사를 고려하는 것이 빠르고 적절한 치료로 이어질 수 있을 것이다.