무도증(chorea)은 불규칙한 근육 수축이 연속적으로 일어나면서 예측할 수 없는 불수의적인 움직임이 주변 부위로 전파되는 양상의 이상운동이다. 서로 다른 원인에 의하여 발생하더라도 그 임상양상은 비슷하기 때문에, 원인질환의 감별에는 임상 정보나 검사결과가 중요한 역할을 하게 된다[1].
무도증은 감염, 자가면역질환, 유전자 변이, 신경퇴행성질환, 뇌졸중, 종양, 약물, 대사성질환 등의 다양한 원인으로 발생하지만, 신경이완제나 레보도파의 장기적인 투약이나 헌팅톤씨병이 가장 흔한 원인이다[2]. 무도증은 매우 드물게 종양과 관련된 신생물딸림 신경학증후군(paraneoplastic neurologic syndrome, PNS)에 의하여 발생할 수 있으며[3], 저자들은 무도증을 초기 증상으로 내원한 환자에서 폐암과 관련된 PNS 증례를 경험하였기에 문헌고찰과 함께 보고한다.
증 례
75세 여자가 양쪽 다리가 따끔거리면서 저리기 시작하였고, 점차 심해지면서 양쪽 다리에 힘이 없고 뻣뻣한 느낌이 들었다. 환자는 20년 전부터 고혈압, 천식으로 다른 의료기관에서 doxofylline, montelukast, felodipine, hydrochlorothiazide를 복용 중이었고, 7년 전부터 손떨림으로 trihexyphenidyl 2 mg을 하루 두 번, 5년 전부터는 pramipexole 0.125 mg을 하루 두 번 복용하기 시작하였다. 환자는 증상 발생 10일 후에 근처 병원에 입원하여 보존적인 치료를 받았으나 이후 10일이 경과하여도 증상이 더 심해졌다고 하였다. 허리 및 엉치 X선검사 및 자기공명영상검사에서는 3-5번째 요추간판탈출 및 척추관협착이 발견되었다. 증상 발생 3주째에 양쪽 다리의 강직과 불수의적 움직임이 발생하여 본 병원으로 전원되었다.
전원 당시 활력징후는 정상이었고 신경학적 진찰에서 양쪽 하지의 근력은 Medical Research Council 4등급으로 저하되어 있었으나, 감각이상은 없었고 바빈스키징후는 음성이었다. 환자는 양쪽 하지 전체에서 지속적이고 불규칙적인 비틀림 운동을 보였고 중등도의 강직이 있었으나, 양쪽 상지에서는 이상운동이 보이지 않았다. 손과 발에서 운동 완만은 없었으며, 동작을 수행할 때 다른 신체 부위에서 이상운동이 증가하거나 양상이 변하지 않았다(Supplementary video).
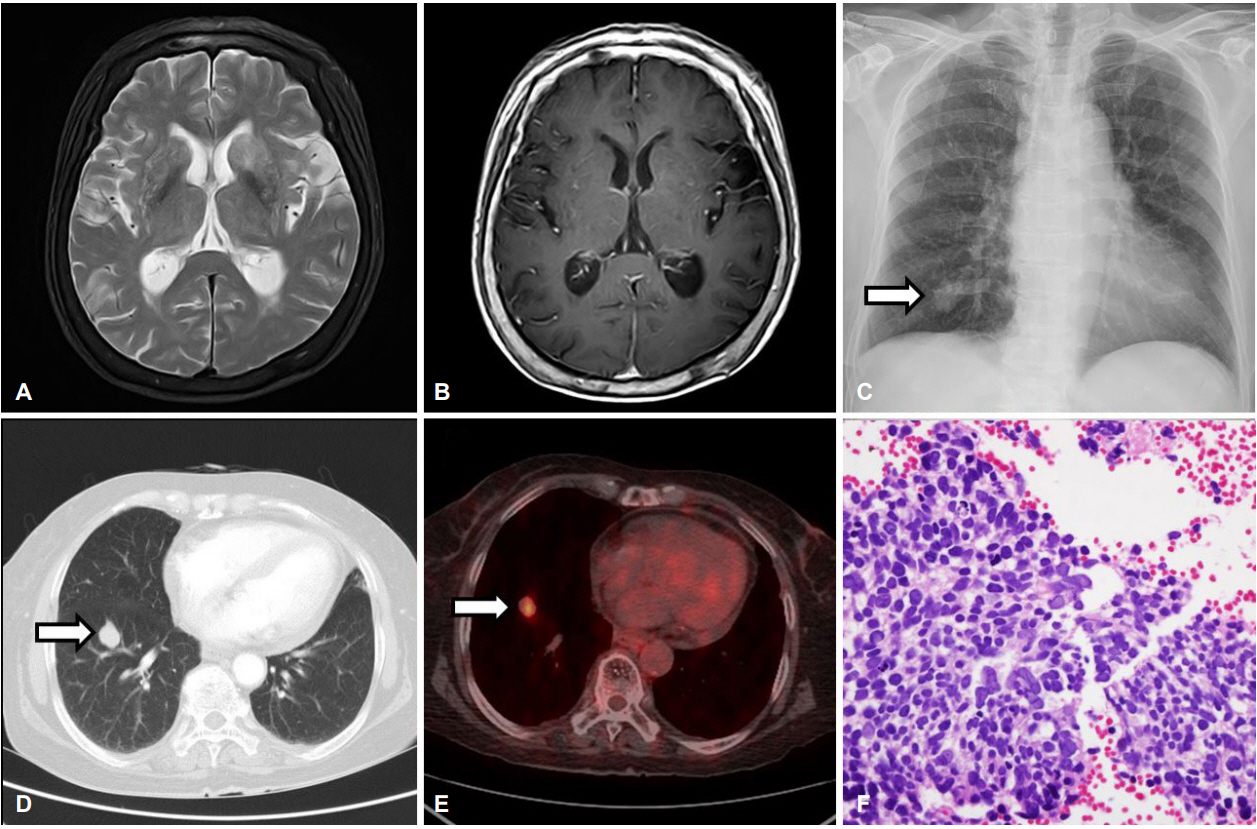
뇌 자기공명영상에서는 소혈관질환 외에 다른 이상은 없었다(Fig. A, B). HTT 유전자검사에서도 CAG 반복수 17/17로 정상범위였다. 혈청 구리는 108.7 μg/dL (75.0-145.0), 24시간 소변 구리 101.3 μg/day (15.0-60.0), 혈청 세룰로플라스민 28.0 mg/dL (17.9-53.3)였다. 당화혈색소는 6.0%, 혈청 포도당은 96 mg/dL (70-110)였다. 뇌척수액검사에서 백혈구 2/μL, 적혈구 0/μL, 단백질 81.8 mg/dL (15-45), 포도당 64 mg/dL (40-80)였고, 혈청 항핵항체, 항카디오리핀항체, 항중성구세포질항체, 류마티스인자는 음성이었고, 갑상선기능검사도 정상이었다.
양측 하지에서 시행한 신경전도검사, 근전도검사는 정상이었으나 감각유발전위검사에서 양측 정중신경감각유발전위 및 양측 뒤 정강신경 감각유발전위에서 중추성 전도장애가 관찰되었다. 이에 다시 검사한 척수 자기공명영상에서 경추 및 요추부 척추관협착이 관찰되었다.
전원된 날부터 자가약인 trihexyphenidyl, pramipexole을 중단하고 지켜보았으나 양쪽 하지의 무도증은 점차 심해졌다. 11일째에 환자는 양손에 힘이 없어 휴대폰을 다룰 수 없다는 증상을 호소하였고, 양손에서도 느린 비틀림운동(athetosis)이 관찰되었다.
한편, 입원 중 정기적으로 시행하던 가슴 X선 추적검사에서 우측 아래 폐에서 방사선비투과음영이 발견되었고(Fig. C), 흉부 전산화단층촬영(Fig. D)과 전신 양전자방출단층촬영(Fig. E)에서 폐암이 의심되었다. 기관지폐포세척검사 및 기관지내시경을 이용한 조직검사에서 소세포폐암이 진단되었다(Fig. F). 신생물딸림 항체검사에서는 항Hu항체 양성이 보고되었다. 호흡기내과로 전과되어 1차 항암 치료를 진행하였으나 무도증은 호전되지 않았고, 2차 항암 치료 예정이었으나 폐렴이 발생하여 증상 발생 8주 후, 폐암 진단 18일 후에 사망하였다.
고 찰
PNS는 신경계를 침범한 신생물딸림 증후군으로, 대부분은 면역기전에 의하여 발생한다[4]. 정상적으로는 신경계에서만 발현되어야 하는 항원이 종양세포에서 발현되는데 이를 종양신경항원이라고 부른다. 종양의 성장을 억제시키기 위하여 인체의 면역체계에서 이 항원에 대하여 면역학적인 공격을 하게 되고[4,5], 이 과정에서 같은 항원을 가지고 있는 신경계 세포들이 손상되면서 나타나는 질환이다. 항원의 위치에 따라 분류하는데, 세포내 항원에 대한 종양신경항체로는 항Hu항체, 항CRMP5/CV2항체, 항Yo항체가 있고, 세포 표면에 결합하는 종양신경항체로는 항VGCC항체, 항NMDAR항체 등이 있다[6,7]. PNS를 일으키는 가장 흔한 종양은 소세포폐암이고, 난소 종양, 유방암, 비소세포폐암 순서이며, 종양신경항체로는 항Hu항체, 항Yo항체가 가장 흔하다[3]. 무도증으로 발현되는 PNS에서는 소세포폐암이 가장 흔한 원인이며, 종양신경항체로는 항CV2/CRMP5항체가 가장 흔하고, 본 증례와 같은 항Hu항체가 다음으로 흔한 항체이다[3].
PNS는 종양 환자의 0.01%에서만 관찰되는 드문 질환으로, 뇌, 척추, 말초신경, 신경근이음부, 근육에 이르기까지 다양한 신경계에 영향을 미칠 수 있어 임상양상, 징후가 다양하다. 대개 종양이 진단되기 이전에 신경학적 증상이 발현되는데, 80%에 달하는 PNS 환자에서 종양이 진단되기 수 개월에서 수 년 전에 PNS 증상이 시작된다[5]. 현재까지는 종양 자체를 치료하는 것이 가장 중요한 치료이며, 면역조절 치료는 그 효과가 불분명하고 일관되지 않다[4,5]. 치료에도 불구하고 예후는 나빠서 운동질환으로 발현되는 환자의 생존율은 16-18개월로 알려져 있다[3].
PNS 자체도 빈도가 낮지만, 그중에서도 무도증으로 발현되는 경우는 굉장히 드물다. 한 등록 연구에서는 전체 1,000예의 PNS 중 11예(1.1%)에서만 무도증을 보였다고 한다[3]. 이 환자들 모두 40세 이상이었고 평균 나이는 65세였는데, 이는 시덴함무도증(Sydenham’s chorea), 헌팅톤병(Huntington’s disease) 등 잘 알려진 다른 무도증보다 발병 연령이 높은 편이다. 무도증의 양상은 다른 원인질환의 그것과 다르지 않았으나, 다른 무도증과는 달리 아급성(subacute)의 경과를 보였다[3]. 따라서, 고령에서 발생한 아급성 경과의 무도증에서는 다른 원인이 명확하지 않다면 PNS 가능성을 고려하여 종양에 대한 탐색을 해야 한다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement
Supplement Print
Print