선천근섬유형불균형(congenital fiber type disproportion, CFTD)은 선천근육병의 드문 아형으로 임상적으로 출생 시부터 경미한 근긴장저하에서 호흡부전을 동반한 근력저하까지 다양하게 나타날 수 있고 유전적으로는 가장 흔한 원인으로 알려진 tropomyosin 3 (TPM3)를 비롯하여 ACTA1, RYR1, SEPN1 등 여러 유전자들과 연관된 이질적인 질환이다[1-3]. 과거 연구에 따르면 TPM3 돌연변이로 인한 선천성 근육병의 50% 이상이 CFTD였고 나머지는 네말린근육병과 캡근육병이었다[4]. TPM3 돌연변이로 인한 CFTD 환자들은 일반적으로 근위부의 근력저하를 보였으며 경부 굴근의 근력저하와 경증의 얼굴 근육 위약 그리고 눈꺼풀처짐 등이 관찰되었다[1,3,4]. 성인이 된 후에 보행이 가능할 정도로 사지 근력이 보존되어 있어도 호흡근이 침범되어 호흡기능 저하가 발생하는 경우가 보고되었지만[1,4] 성인기에 호흡부전이 발생하여 선천근육병으로 진단되는 것은 드문 일이다. 저자들은 성인기까지 사지 근력이 상대적으로 보존되어 있어 호흡부전이 발생한 후에야 TPM3 유전자 돌연변이로 인한 CFTD로 진단한 가계를 경험하였기에 이를 보고하고자 한다.
증 례
증례1
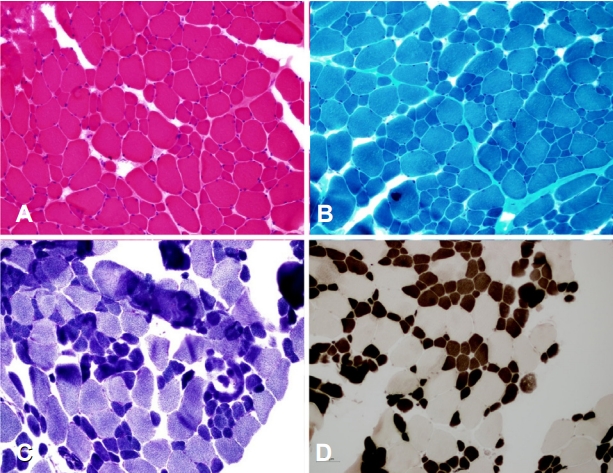
22세 여자가 일주일 전부터 기침, 가래 등 상기도 감염의 징후가 있었으며 3일 전부터는 숨이 차고 자꾸 졸려 하였고 병원 방문 당일 저녁에 자극에 대한 반응이 떨어지고 처지는 모습이 관찰되어 타 병원 응급실을 방문하였다. 동맥혈가스분석(arterial blood gas analysis)에서 pH 7.2, PaO2 70 mmHg 및 PaCO2 85 mmHg로 CO2 저류와 의식저하를 보여 기관내삽관 후 중환자실로 입실하여 인공호흡 치료를 시작하였다. 의식은 정상수준을 회복하였으나 삽관 제거 후에도 지속적으로 CO2 저류 소견이 관찰되어 비침습양압환기(noninvasive positive pressure ventilation, NIPPV)를 적용하였다. 당시 심전도검사는 정상이었고, 경흉부 심초음파검사에서 좌심실 박출률(ejection fraction, 67%)은 정상이었으며 다른 구조 이상은 관찰되지 않았다. 호흡기능검사에서 강제폐활량(forced vital capacity, FVC)이 1.54 L (predicted value 32%)로 낮게 측정되었다. 환자는 신생아 시기에 전반적인 근력저하와 경도의 운동발달 지연이 있었으나, 병원에 오기 전까지 일상생활에 불편함이 없고 걷거나 계단을 오르는 데에 어려움이 없었으며 달리기도 가능하였다. 환자의 가족 중에서 어머니와 오빠도 근력저하가 있었고 오빠는 환자와 비슷한 병력을 가지고 있었다(Fig. 1-A). 신경학적 진찰에서 환자는 근육병얼굴과 높은 구개, 눈꺼풀처짐, 전반적인 근위축 그리고 요추전만증이 있었다. 도수근력검사에서 경부 굴근과 양측 상지 근위부 근육이 Medical Research Council (MRC) 4+등급으로 측정되었고, 양측 하지 근육은 5등급이었다. 심부건반사는 사지에서 모두 소실되었으며 병적반사는 관찰되지 않았다. 혈청 크레아틴키나아제는 57 IU/L (정상치, 5-217 IU/L)로 정상이었다. 신경전도검사는 정상이었으며 침근전도검사에서 전반적으로 진폭이 낮고 지속 시간이 짧은 다상운동단위활동전위가 관찰되어 근육병에 합당한 결과였다. 환자의 좌측 가쪽넓은근(vastus lateralis)에서 시행한 근생검에서는 헤마톡실린-에오신 염색에서 근세포 크기의 다양성이 증가되어 있었으며, ATPase (pH 4.3) 염색에서 1형 근섬유가 수적으로 우세하게 관찰되었고 1형 근섬유의 크기가 2형 근섬유에 비하여 60% 이상 작았다. 변형 고모리-트리크롬 염색에서 캡 구조(cap structure)나 네말린 소체(nemalin bodies)는 관찰되지 않았다(Fig. 2). 임상양상과 근생검 결과를 고려하여 CFTD에 합당하다고 판단하였고 표적 차세대염기서열분석법(next-generation sequencing, NGS)으로 TPM3 유전자에서 이형접합 과오돌연변이인 c.502C>T (p.Arg168Cys, reference sequence NM_152263.2)가 발견되었다(Fig. 1-B). 이 과오돌연변이는 CFTD 이외에도 캡근육병과 네말린근육병에서 발견된 보고가 있었다[4,5]. 저자들은 TPM3 유전자 돌연변이로 인한 CFTD로 진단하였고, 어머니에게도 유전자 검사를 권유하였으나 거부하여 시행하지 못하였다. 그렇지만 환자의 어머니 또한 환자와 얼굴이 비슷하였고 경부 굴근의 근력저하가 있었으며 보행장애는 없었으나 계단을 오르거나 달리기를 잘 하지 못하였기 때문에 임상양상으로 보아 어머니도 이환되었을 것으로 추정하였고 상염색체 우성 유전형 가계로 판단하였다. 환자는 5년이 지난 현재까지 NIPPV를 야간에만 적용하고 있으며 보행 가능한 상태를 유지하고 있다.
증례2 (증례1의 오빠)
26세 남자가 일주일 전부터 가슴 답답함과 복부 팽만감을 호소하였고 야간 수면 중 호흡정지 상태로 발견되어 CPCR 시행 후 타 병원 응급실로 이송되었다. 기관내삽관 후 중환자실에서 인공호흡 치료를 받았으며, 의식은 정상수준을 회복하였지만 호흡기능 저하로 기관 절개술 후 NIPPV를 시작하였다. 경흉부 심초음파에서 좌심실 박출률(63%)은 정상수준이었고 국소벽 운동이상도 관찰되지 않았으나 우심실의 크기가 커져 있었다. 호흡기능검사에서 FVC가 1.41 L (predicted value 27%)로 낮게 측정되었다. 환자는 출생 시 근긴장저하를 보였고 운동발달 지연으로 생후 24개월부터 걷기 시작하였으며 달리기를 하면 남들보다 더 힘이 들었고 꼴찌를 하곤 하였다. 16세에 특발척추측만증으로 수술을 받고 합병증 없이 회복하였고 일주일 전까지는 일상 활동에 문제가 없었다고 하였다. 환자는 여동생과 마찬가지로 근육병얼굴과 높은 구개, 눈꺼풀처짐, 아래턱후퇴가 있었고 발목관절 구축과 현저한 요추전만증이 관찰되었다. 전반적으로 근위축이 있었으며 도수근력검사에서 경부 굴근과 발등굽힘근에서 MRC 3등급으로 가장 심한 근력 저하가 관찰되었다. 혈청 크레아틴키나아제는 39 IU/L (정상치, 5-217 IU/L)로 정상이었다. 침근전도검사는 근육병에 합당하였으며 NGS를 시행하여 여동생과 같은 TPM3 유전자의 이형접합 과오돌연변이를 확인하였다. 환자는 NIPPV 시행 후 10년 이상 야간 호흡부전의 재발은 없었으며 5분 이상 독립보행이 가능한 상태로 호흡 재활 치료를 받고 있다.
고 찰
CFTD는 조직학적으로 2형 근섬유에 비하여 1형 근섬유가 위축되어 있는 것이 특징적이며, 다른 유의한 구조 이상 없이 1형 근섬유의 크기가 2형 근섬유보다 최소 12% 이상 작으며 임상적으로 선천근육병에 부합하면 CFTD로 진단할 수 있다[2]. 현재까지 CFTD와의 연관성이 가장 잘 정립되어 있는 원인 유전자들은 TPM3, RYR1 그리고 ACTA1 유전자이며 TPM3 유전자가 가장 흔한 원인으로 알려져 있다[3]. 하지만 아직까지 국내에서는 TPM3 유전자 돌연변이로 인한 CFTD에 대한 보고는 없었다[6]. TPM3 유전자 돌연변이에서 근섬유 위축이 1형 근섬유에만 발생하는 것은 TPM3 유전자가 1형 근섬유에 있는 slow type의 알파-트로포미오신(α-tropomyosinslow, α-TPMslow)을 암호화하기 때문이다[1,7]. 반면에 fast type의 알파-트로포미오신(α-tropomyosinfast, α-TPMfast)은 2형 근섬유에 있으며 TPM1 유전자가 암호화한다. Clarke 등[1]이 보고한 바에 따르면, TPM3 유전자 돌연변이에 의한 CFTD 환자들은 일반적으로 출생시나 신생아 시기 이후부터 경증에서 중등도의 근력저하가 있었고, 가장 흔하게 침범하는 근육들은 경부 굴근과 신전근, 척추주위근, 복근을 포함한 몸통의 움직임에 관여하는 근육들과 사지의 근위부 근육 그리고 발등굽힘근이었다. 또한 경도의 얼굴 근육 위약과 눈꺼풀처짐도 빈번하게 나타났다. 그리고 보행이 가능한 상태일지라도 야간 호흡곤란이 발생하여 NIPPV를 시행하고 있는 경우도 있었다[1]. 본 증례 환자들도 경부 굴근과 발등굽힘근에서 근력저하가 가장 두드러졌으며, 두 환자 모두 상대적으로 사지의 근력저하가 심하지 않은데도 불구하고 호흡부전으로 인한 의식 저하가 발생하여 인공호흡 보조가 필요하였다. TPM3 돌연변이로 인한 선천근육병에서 호흡근의 근력저하가 발생하는 것은 호흡근이 주로 지속적이고 저강도의 근수축을 담당하는 1형 근섬유로 구성되어 있기 때문이다[7]. 그리고 TPM3 돌연변이가 발현된 α-TPMslow은 정상 α-TPMslow이 매개하는 액틴-미오신 상호작용에 손상을 주기 때문에 근수축력이 저하된다[7,8]. 특히 본 증례를 포함한 TPM3 유전자의 pArg168 잔기(residue)는 (e.g. p.Arg168Cys, p.Arg168Gly, and p.Arg168His) 돌연변이가 잘 발생하는 부분으로 알려져 있다[3]. p.Arg168 잔기가 액틴이 결합하는 부위에 매우 인접해 있기 때문에 이 위치의 돌연변이로 액틴에 대한 α-TPMslow의 친화력이 더욱 감소하여 근력저하가 잘 발생한다는 기전이 제시된 바 있다[7,8]. 현재까지 보고된 p.Arg168Cys 과오돌연변이는 대부분 de novo이며 신생아 시기부터 사지 근력저하가 서서히 진행하여 성인기에도 보행이 가능한 경우가 많지만 호흡근의 위약도 같이 진행되면서 인공호흡 보조가 필요하기도 하였다[1,4,5,8]. 본 가계는 두 세대를 포함하는 상염색체 우성유전가계로 한 세대가 모두 호흡곤란으로 인한 의식저하로 선천근육병으로 진단되었고 현재까지 보행은 가능하나 호흡부전이 발생한 이후로는 지속적으로 NIPPV가 필요한 상태이다. 따라서 본 가계와 같이 TPM3 유전자 돌연변이로 인한 선천근육병 환자에서 호흡근의 침범이 환자의 예후에 결정적인 역할을 하기 때문에 조기 진단이 무엇보다 중요하다. TPM3 유전자 돌연변이로 인한 선천근육병이 임상적으로 다양하게 나타날 수 있고 조직병리학적으로도 CFTD를 포함하여 캡근육병이나 네말린근육병으로도 나타날 수 있어서 진단하기가 쉽지 않지만, 흔히 발생하는 근력저하의 양상이나 예후에 대한 다양한 보고들이 본 증례와 더불어 임상에서 환자를 다루는 데 도움이 될 것이다. TPM3 유전자 돌연변이로 인한 CFTD에서 성인이 된 후로도 보행에 지장이 없을 만큼 근력이 충분하게 남아 있을지라도 호흡부전이 발생하여 환자에게 치명적인 결과를 초래할 수 있기 때문에 잠재적인 호흡기능 저하를 조기에 발견하기 위하여 호흡기능에 대한 지속적인 감시와 평가가 필요하다.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print